Tartaric Acid Amide Derivative and Method of Producing the Same
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Amide, and Paratoluenesulfonamide on the Amide of Silver,On the Imides
68 CHEMISTRY: E. C. FRANKLIN METALLIC SALTS OF AMMONO ACIDS By Edward C. Franklin DEPARTMENT OF CHEMISTRY, STANFORD UNIVERSITY Presented to the Academy, January 9. 1915 The Action of Liquid-Ammonia Solutions of Ammono Acids on Metallic Amides, Imides, and Nitrides. The acid amides and imides, and the metallic derivatives of the acid amides and imides are the acds, bases, and salts respectively of an ammonia system of acids, bases, and salts.1 Guided by the relationships implied in the above statement Franklin and Stafford were able to prepare potassium derivatives of a considerable number of acid amides by the action of potassium amide on certain acid amides in solution in liquid ammonia. That is to say, an ammono base, potassium amide, was found to react with ammono acids in liquid ammonia to form ammono salts just as the aquo base, potassium hydrox- ide, acts upon aquo acids in water solution to form aquo salts. Choos- ing, for example, benzamide and benzoic acid as representative acids of the two systems, the analogous reactions taking place respectively in liquid ammonia and water are represented by the equations: CH6CONH2+KNH2 = C6H5CONHK + NHs. CseHCONH2 + 2KNH2 = CIHsCONK2 + 2NH3. CH6tCOOH + KOH = CIH6COOK + H2O. The ammono acid, since it is dibasic, reacts with either one or two molecules of potassium amide to form an acid and a neutral salt. Having thus demonstrated the possibility of preparing ammono salts of potassium by the interaction of potassium amide and acid amides in liquid ammonia solution, it was further found that ammono salts of the heavy metals may be prepared by the action of liquid ammonia solutions of ammono acids on insoluble metallic amides, imides, and nitrides-that is, by reactions which are analogous to the formation of aquo salts in water by the action of potassium hydroxide on insoluble metallic hydroxides and oxides. -

Amide Activation: an Emerging Tool for Chemoselective Synthesis
Featuring work from the research group of Professor As featured in: Nuno Maulide, University of Vienna, Vienna, Austria Amide activation: an emerging tool for chemoselective synthesis Let them stand out of the crowd – Amide activation enables the chemoselective modification of a large variety of molecules while leaving many other functional groups untouched, making it attractive for the synthesis of sophisticated targets. This issue features a review on this emerging field and its application in total synthesis. See Nuno Maulide et al., Chem. Soc. Rev., 2018, 47, 7899. rsc.li/chem-soc-rev Registered charity number: 207890 Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue Amide activation: an emerging tool for chemoselective synthesis Cite this: Chem. Soc. Rev., 2018, 47,7899 Daniel Kaiser, Adriano Bauer, Miran Lemmerer and Nuno Maulide * It is textbook knowledge that carboxamides benefit from increased stabilisation of the electrophilic carbonyl carbon when compared to other carbonyl and carboxyl derivatives. This results in a considerably reduced reactivity towards nucleophiles. Accordingly, a perception has been developed of amides as significantly less useful functional handles than their ester and acid chloride counterparts. Received 27th April 2018 However, a significant body of research on the selective activation of amides to achieve powerful DOI: 10.1039/c8cs00335a transformations under mild conditions has emerged over the past decades. This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product rsc.li/chem-soc-rev synthesis, highlighting the synthetic applications and the potential of this approach. Creative Commons Attribution 3.0 Unported Licence. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -
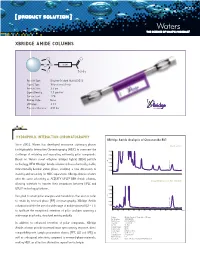
Xbridge Amide Columns
[ PRODUCT SOLUTION ] XBRIDGE AMIDE COLUMNS O O O Si Linker O NH 2 Particle Type: Ethylene Bridged Hybrid [BEH] Ligand Type: Trifunctional Amide Particle Size: 3.5 µm Ligand Density: 7.5 µmol/m2 Carbon Load: 12% Endcap Style: None pH Range: 2-11 Pressure Tolerance: 400 bar HYDROPHILIC INTERacTION CHROMATOGRAPHY XBridge Amide Analysis of Ginsenoside Rb1 Since 2003, Waters has developed innovative stationary phases Orento extract for Hydrophilic Interaction Chromatography [HILIC] to overcome the 0.010 challenge of retaining and separating extremely polar compounds. 0.008 Based on Waters novel ethylene bridged hybrid [BEH] particle 0.006 AU technology, NEW XBridge™ Amide columns utilize a chemically stable, 0.004 0.002 Ginsenoside Rb1 trifunctionally-bonded amide phase, enabling a new dimension in 0.000 stability and versatility for HILIC separations. XBridge Amide columns offer the same selectivity as ACQUITY UPLC® BEH Amide columns, 20 µg/mL Ginsenoside Rb1 standard allowing scientists to transfer their separations between HPLC and 0.010 ® UPLC technology platforms. 0.008 0.006 Designed to retain polar analytes and metabolites that are too polar AU 0.004 to retain by reversed-phase [RP] chromatography, XBridge Amide 0.002 columns facilitate the use of a wide range of mobile phase pH [2 – 11] 0.000 Ginsenoside Rb1 to facilitate the exceptional retention of polar analytes spanning a 0.00. 5 1.01. 5 2.02. 5 3.03. 5 4.04. 5 5.05. 5 6.06. 5 min wide range in polarity, structural moiety and pKa. Column: XBridge Amide, 3.5 µm, 4.6 x 150 mm Part Number: 186004869 Mobile Phase : 80/20 ACN/H2O In addition to enhanced retention of polar compounds, XBridge Flow Rate: 1.4 mL/min Inj. -

In This Handout, All of Our Functional Groups Are Presented As Condensed Line Formulas, 2D and 3D Formulas and with Nomenclature Prefixes and Suffixes (If Present)
In this handout, all of our functional groups are presented as condensed line formulas, 2D and 3D formulas and with nomenclature prefixes and suffixes (if present). Organic names are built on a foundation of alkanes, alkenes and alkynes. Those examples are presented first and you need to know those rules. The strategies can be found in Chapter 4 of our textbook (alkanes: pages 93-98, cycloalkanes 102-104, alkenes: pages 104-110, alkynes: pages 112-113 and combinations of all of them 113-115). After introducing examples of alkanes, alkenes, alkynes and combinations of them, the functional groups are presented in order of priority. A few nomenclature examples are provided for each of the functional groups. Examples of the various functional groups are presented on pages 115-135 in the textbook. Two overview pages are on pages 136-137. Some functional groups have a suffix name when they are the highest priority functional group and a prefix name when they are not the highest priority group, and these are added to the skeletal names with identifying numbers and stereochemistry terms (E and Z for alkenes, R and S for chiral centers and cis and trans for rings). Several low priority functional groups only have a prefix name. A few additional special patterns are shown on pages 98-102. The only way to learn this topic is practice (over and over). The best practice approach is to actually write out the names (on an extra piece of paper or on a white board, and then do it again). The same functional groups are used throughout the entire course. -

Detection of Atmospheric Gaseous Amines and Amides by a High-Resolution Time-Of-flight Chemical Ionization Mass Spectrometer with Protonated Ethanol Reagent Ions
Atmos. Chem. Phys., 16, 14527–14543, 2016 www.atmos-chem-phys.net/16/14527/2016/ doi:10.5194/acp-16-14527-2016 © Author(s) 2016. CC Attribution 3.0 License. Detection of atmospheric gaseous amines and amides by a high-resolution time-of-flight chemical ionization mass spectrometer with protonated ethanol reagent ions Lei Yao1, Ming-Yi Wang1,a, Xin-Ke Wang1, Yi-Jun Liu1,b, Hang-Fei Chen1, Jun Zheng2, Wei Nie3,4, Ai-Jun Ding3,4, Fu-Hai Geng5, Dong-Fang Wang6, Jian-Min Chen1, Douglas R. Worsnop7, and Lin Wang1,4 1Shanghai Key Laboratory of Atmospheric Particle Pollution and Prevention (LAP3), Department of Environmental Science & Engineering, Fudan University, Shanghai 200433, China 2Jiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, Nanjing University of Information Science & Technology, Nanjing 210044, China 3Joint International Research Laboratory of Atmospheric and Earth System Sciences, School of Atmospheric Science, Nanjing University, Nanjing 210023, China 4Collaborative Innovation Center of Climate Change, Nanjing 210023, China 5Shanghai Meteorology Bureau, Shanghai 200135, China 6Shanghai Environmental Monitoring Center, Shanghai 200030, China 7Aerodyne Research, Billerica, MA 01821, USA anow at: Center for Atmospheric Particle Studies, Carnegie Mellon University, Pittsburgh, PA 15213, USA bnow at: Pratt School of Engineering, Duke University, Durham, NC 27705, USA Correspondence to: Lin Wang([email protected]) Received: 7 June 2016 – Published in Atmos. Chem. Phys. Discuss.: 22 June 2016 Revised: 15 October 2016 – Accepted: 3 November 2016 – Published: 23 November 2016 Abstract. Amines and amides are important atmospheric concentrations of amines ranged from a few parts per trillion organic-nitrogen compounds but high time resolution, highly by volume to hundreds of parts per trillion by volume, con- sensitive, and simultaneous ambient measurements of these centrations of amides varied from tens of parts per trillion by species are rather sparse. -

COMMUNICATION Catalytic Ester and Amide To
Catalytic Ester and Amide to Amine Interconversion: Nickel-Catalyzed Decarbonylative Amination of Esters and Amides by C−O and C−C Bond Activation Item Type Article Authors Yue, Huifeng; Guo, Lin; Liao, Hsuan-Hung; Cai, Yunfei; Zhu, Chen; Rueping, Magnus Citation Yue H, Guo L, Liao H-H, Cai Y, Zhu C, et al. (2017) Catalytic Ester and Amide to Amine Interconversion: Nickel-Catalyzed Decarbonylative Amination of Esters and Amides by C−O and C −C Bond Activation. Angewandte Chemie International Edition 56: 4282–4285. Available: http://dx.doi.org/10.1002/anie.201611819. Eprint version Post-print DOI 10.1002/anie.201611819 Publisher Wiley Journal Angewandte Chemie International Edition Rights This is the peer reviewed version of the following article: Catalytic Ester and Amide to Amine Interconversion: Nickel-Catalyzed Decarbonylative Amination of Esters and Amides by C-O and C-C Bond Activation, which has been published in final form at http:// doi.org/10.1002/anie.201611819. This article may be used for non-commercial purposes in accordance With Wiley Terms and Conditions for self-archiving. Download date 23/09/2021 09:53:43 Link to Item http://hdl.handle.net/10754/623218 COMMUNICATION Catalytic Ester and Amide to Amine Interconversion: Nickel- Catalyzed Decarbonylative Amination of Esters and Amides via C- O and C-C Bond Activation Huifeng Yue[a], Lin Guo[a], Hsuan-Hung Liao[a], Yunfei Cai[a], Chen Zhu[a], and Magnus Rueping[a,b]* Abstract: An efficient nickel catalyzed decarbonylative amination reaction of aryl and heteroaryl esters has been achieved for the first a) Schmidt reaction; Curtius, Hoffmann, Lossen rearrangement time. -

Trifluoromethanesulfonic Anhydride in Amide Activation:A Powerful Tool to Forge Heterocyclic Cores |
TCIMAIL No. 186 l Spring 2021 Spring 2021 l No. 186 TCIMAIL Research Article Trifluoromethanesulfonic Anhydride in Amide Activation: A Powerful Tool to Forge Heterocyclic Cores Haoqi Zhang, Margaux Riomet, Nuno Maulide Institute of Organic Chemistry, University of Vienna, Währinger Strasse 38, 1090 Vienna, Austria E-mail: [email protected] Abstract Trifluoromethanesulfonic anhydride is a powerful reagent for the activation of a wide range of functionalities. Among them, the electrophilic activation of amides is a compelling approach to access heterocycles under mild reaction conditions. Herein, we outline the most recent achievements in the construction of heterocycles via amide activation with trifluoromethanesulfonic anhydride. Keywords: Trifluoromethanesulfonic anhydride, triflic anhydride, amide activation, heterocycle, Umpolung Introduction well as the presence (or absence) of a base (Scheme 1). When subjected to triflic anhydride 2, amides are In recent decades, trifluoromethanesulfonic generally rapidly converted to the corresponding anhydride, also known as triflic anhydride, has proven iminium triflates 3. Further deprotonation, usually by to be an extraordinary reagent for a broad range a pyridine derivative can lead to formation of a highly of transformations. Given its high affinity towards electrophilic intermediate – a nitrilium ion 5 in case of O-nucleophiles, reaction with alcohols, carbonyls, sulfur- secondary amides, or a keteniminium ion 6 for their phosphorus- and iodine oxides towards formation of tertiary counterparts. -

Use of Chemically Modified Poly(Ethylene Terephthalate)-G- (Acryl Amide) Fibers for Α-Amylase Immobilization
http://www.e-polymers.org e-Polymers 2007, no. 070 ISSN 1618-7229 Use of chemically modified poly(ethylene terephthalate)-g- (acryl amide) fibers for α-amylase immobilization Zülfikar Temoçin, Mustafa Yiğitoğlu* Kırıkkale University, Science and Arts Faculty, Chemistry Department, Yahşihan, 71450 Kırıkkale, Turkey; Fax: +90-318-3572461; [email protected]. (Received: 12 April, 2007; published: 30 June, 2007) Abstract: Acryl amide grafted Poly(ethylene terephthalate) (AAm-g-PET) fiber was used for covalent coupling of α-amylase. The amide groups of Poly(acryl amide) were converted to the amine groups by Hofmann degradation reaction. The amine groups were activated by glutaraldehyde, before coupling of the enzyme. The free α-amylase and immobilized α-amylase were characterized by determining the activity profile as function of pH, temperature, thermal stability and storage stability. For the immobilized α-amylase, operational stability was also determined. The immobilization of α-amylase on support caused the optimal reaction pH to shift from 5 to 6. The maximum activity of the free and immobilized enzymes occurred at 0 50 C. Km for the immobilized system was higher than that for the free enzyme. The activity of the free enzyme ended in 30 days, whereas the activity of the immobilized enzyme lasted for 60 days at storage conditions. α-Amylase immobilized on matrix maintained 40% of its original activity after 30 times of repeated use. Introduction α-Amylase is a kind of starch-hydrolyzed enzyme. It is an endo-enzyme that can cut α-1,4 glucosidic linkage of starch molecules randomly to form oligo with different molecular weight such as glucose, cane sugar and straight chain oligo, rapidly reducing the viscosity of liquid and has been used in big volume starch hydrolysis industry [1]. -

1 Chapter 3: Organic Compounds: Alkanes and Cycloalkanes
Chapter 3: Organic Compounds: Alkanes and Cycloalkanes >11 million organic compounds which are classified into families according to structure and reactivity Functional Group (FG): group of atoms which are part of a large molecule that have characteristic chemical behavior. FG’s behave similarly in every molecule they are part of. The chemistry of the organic molecule is defined by the function groups it contains 1 C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic C N C C C X X= F, Cl, Br, I amines Alkenes Alkyl Halide H C C C O C C O Alkynes alcohols ethers acidic H H H C S C C C C S C C H sulfides C C thiols (disulfides) H H Arenes Carbonyl-oxygen double bonds (carbonyls) Carbon-nitrogen multiple bonds acidic basic O O O N H C H C O C Cl imine (Schiff base) aldehyde carboxylic acid acid chloride O O O O C C N C C C C O O C C nitrile (cyano group) ketones ester anhydrides O C N amide opsin Lys-NH2 + Lys- opsin H O H N rhodopsin H 2 Alkanes and Alkane Isomers Alkanes: organic compounds with only C-C and C-H single (s) bonds. general formula for alkanes: CnH(2n+2) Saturated hydrocarbons Hydrocarbons: contains only carbon and hydrogen Saturated" contains only single bonds Isomers: compounds with the same chemical formula, but different arrangement of atoms Constitutional isomer: have different connectivities (not limited to alkanes) C H O C4H10 C5H12 2 6 O OH butanol diethyl ether straight-chain or normal hydrocarbons branched hydrocarbons n-butane n-pentane Systematic Nomenclature (IUPAC System) Prefix-Parent-Suffix -

Downloaded 9/24/2021 4:45:01 AM
ORGANIC CHEMISTRY FRONTIERS View Article Online REVIEW View Journal | View Issue Recent developments in dehydration of primary amides to nitriles Cite this: Org. Chem. Front., 2020, 7, 3792 Muthupandian Ganesan *a and Paramathevar Nagaraaj *b Dehydration of amides is an efficient, clean and fundamental route for the syntheses of nitriles in organic chemistry. The two imperative functional groups viz., amide and nitrile groups have been extensively dis- cussed in the literature. However the recent development in the century-old dehydration method for the conversion of amides to nitriles has hardly been reported in one place, except a lone review article which dealt with only metal catalysed conversions. The present review provides broad and rapid information on the different methods available for the nitrile synthesis through dehydration of amides. The review article Received 15th July 2020, has major focus on (i) non-catalyzed dehydrations using chemical reagents, and (ii) catalyzed dehy- Accepted 19th September 2020 drations of amides using transition metal, non-transition metal, organo- and photo-catalysts to form the DOI: 10.1039/d0qo00843e corresponding nitriles. Also, catalyzed dehydrations in the presence of acetonitrile and silyl compounds as rsc.li/frontiers-organic dehydrating agents are highlighted. 1. Introduction polymers, materials, etc.1 Examples of pharmaceuticals contain- ing nitrile groups include vildagliptin, an anti-diabetic drug2 Nitriles are naturally found in various bacteria, fungi, plants and anastrazole, a drug -

Carboxylic Acids a Carbonyl with One OH Attached Is Called a Carboxylic
Carboxylic Acids A carbonyl with one OH attached is called a carboxylic acid One important property of carboxylic acids is the acidity O O pKa = 4.7 B BH H3C OH H3C O acetic acid Upon deprotonation a carboxylate is formed Nomenclature There are two important guidelines to know about carboxylic acids: 1) The carboxylic acid has the highest priority in naming 2) In common names, the point of substitution is labeled by the Greek letter counting from the carbonyl " O OH # ! This naming is common practice amongst organic chemists, e,g, substitution at α-carbon Examples (E)-2-pentenoic acid 3-bromo-2-methylpentanoic acid Or β-bromo-α-methylpentanoic acid (common) Common Names Many acid compounds have a common name Most prevalent amongst these are aromatic compounds Physical Properties Most physical properties of carboxylic acids are a result of hydrogen bonding The carboxylic acids form dimers through hydrogen bonding This hydrogen bonding causes a higher melting point and boiling point compared to compounds of similar molecular weight Acidity As observed previously, carboxylic acids are far more acidic than alcohols This is due to the stability of the anion formed after deprotonation Can also stabilize anion through inductive effects Polar bonds near anion source can stabilize negative charge (inductive effect) Consider the acetate group again: Carboxylate Salts Upon deprotonation of a carboxylic acid obtain a carboxylate salt This salt has different physical properties than the acid form (similar to the difference in amine salts discussed