Part A. Colorimetric Method for Determination of Barium Part B. the Icrm Oscopic Identification of Lithium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Derivation of Proposed 2007 Draft Matrix Soil Standards for Barium
Derivation of Proposed 2007 Draft Matrix Soil Standards for Barium Glyn R. Fox Environmental Management Branch Environmental Protection Division September 24, 2007 Victoria, British Columbia Table of Contents Page 1. Introduction ……………………………………………………………………… 4 2. Details Related to Derivation of Proposed 2007 Draft Matrix Soil Standards 2.1 Derivation of Human Health Protection Standards – Intake of contaminated soil ………………………………………………………….. 7 2.2 Derivation of Human health Protection Standards – Groundwater used for drinking water ……………………………….…………….......... 7 2.3 Derivation of Environmental Protection Standards – Toxicity to soil invertebrates and plants ……………………………………….......... 9 2.4 Derivation of Environmental Protection Standards – Livestock ingesting soil and fodder ………………………………………………… 9 2.5 Derivation of Environmental Protection Standards – Major microbial function impairment ………………………………….……… 10 2.6 Derivation of Environmental Protection Standards – Groundwater flow to surface water used by aquatic life ..…………………………… 10 2.7 Derivation of Environmental Protection Standards – Groundwater used for livestock watering ……………………………………………… 11 2.8 Derivation of Environmental Protection Standards – Groundwater used for irrigation ………………………………………………………… 12 2.9 CSST “Background” Adjustment ……………………..……………… 12 2.10 Application of CSST Rounding-off Rule ……………………………… 12 3. References …….…………………………………………………………………… 13 2 Table of Contents (continued) Page 4. Exhibits Exhibit 1. Proposed 2007 Draft Matrix Soil Standards for Barium ……..… 19 5. Tables -
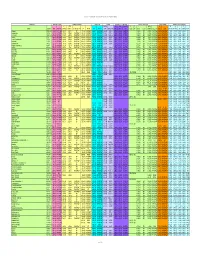
Chemical-Specific Parameters Supporting Table May 2016 Analyte
Regional Screening Level (RSL) Chemical-specific Parameters Supporting Table May 2016 Contaminant Molecular Weight Volatility Parameters Melting Point Density Diffusivity in Air and Water Partition Coefficients Water Solubility Tapwater Dermal Parameters H` (atm- Density Dia Diw Dia and Diw Kd Kd Koc log Kow S B τevent t* Kp 3 3 2 2 Analyte CAS No. MW MW Ref (unitless) m /mole) H` and HLC Ref VP VP Ref MP MP Ref (g/cm ) Density Ref (cm /s) (cm /s) Ref (L/kg) Ref (L/kg) Koc Ref (unitless) log Kow Ref (mg/L) S Ref (unitless) (hr/event) (hr) (cm/hr) K Ref Acephate 30560-19-1 1.8E+02 PHYSPRO 2.0E-11 5.0E-13 EPI 1.7E-06 PHYSPROP 8.8E+01 PHYSPROP 1.4E+00 CRC89 3.7E-02 8.0E-06 WATER9 1.0E+01 EPI -8.5E-01 PHYSPRO 8.2E+05 PHYSPROP 2.1E-04 1.1E+00 2.7E+00 4.0E-05 EPI Acetaldehyde 75-07-0 4.4E+01 PHYSPRO 2.7E-03 6.7E-05 PHYSPROP 9.0E+02 PHYSPROP -1.2E+02 PHYSPROP 7.8E-01 CRC89 1.3E-01 1.4E-05 WATER9 1.0E+00 EPI -3.4E-01 PHYSPRO 1.0E+06 PHYSPROP 1.3E-03 1.9E-01 4.5E-01 5.3E-04 EPI Acetochlor 34256-82-1 2.7E+02 PHYSPRO 9.1E-07 2.2E-08 PHYSPROP 2.8E-05 PHYSPROP 1.1E+01 PubChem 1.1E+00 PubChem 2.2E-02 5.6E-06 WATER9 3.0E+02 EPI 3.0E+00 PHYSPRO 2.2E+02 PHYSPROP 3.1E-02 3.4E+00 8.2E+00 5.0E-03 EPI Acetone 67-64-1 5.8E+01 PHYSPRO 1.4E-03 3.5E-05 PHYSPROP 2.3E+02 PHYSPROP -9.5E+01 PHYSPROP 7.8E-01 CRC89 1.1E-01 1.2E-05 WATER9 2.4E+00 EPI -2.4E-01 PHYSPRO 1.0E+06 PHYSPROP 1.5E-03 2.2E-01 5.3E-01 5.1E-04 EPI Acetone Cyanohydrin 75-86-5 8.5E+01 PHYSPRO 8.1E-08 2.0E-09 PHYSPROP 3.4E-01 PHYSPROP -1.9E+01 PHYSPROP 9.3E-01 CRC89 8.6E-02 1.0E-05 WATER9 1.0E+00 -

Massachusetts Chemical Fact Sheet
Massachusetts Chemical Fact Sheet Hexavalent Chromium Table 1: HEXAVALENT CHROMIUM COMPOUNDS: Compounds SELECTED EXAMPLES* Compound Chemical Formula CAS # This fact sheet is part of a series of chemical fact sheets Ammonium chromate (NH ) Cr0 7788-98-9 developed by TURI to help Massachusetts companies, 4 2 4 community organizations and residents understand the Ammonium dichromate (NH4)2Cr2O7 7789-09-5 chemical’s use and health and environmental effects, as Barium chromate BaCrO4 10294-40-3 well as the availability of safer alternatives. tert-Butyl Chromate [(CH3)3CO]2CrO2 1189-85-1 Hexavalent chromium compounds are a toxic form of Calcium chromate CaCrO4 13765-19-0 chromium and are used in a variety of industrial processes Chromic acid H2CrO4 7738-94-5 and products. Chromium VI chloride CrCl6 14986-48-2 Hexavalent chromium compounds are human carcinogens, Chromic trioxide CrO3 1333-82-0 mutagens and developmental toxicants and are acutely Hexavalent chromium ion Cr6+ 18540-29-9 toxic. Non-hexavalent chromium compounds do not pose Lead chromate PbCrO4 7758-97-6 the same level of concern with regard to either chronic or Lead chromate oxide PbCrO4-PbO 8454-12-1 acute toxicity. Potassium chlorochromate KCrO3Cl 16037-50-6 Until 2011, all chromium compounds were treated as Potassium chromate K2CrO4 7789-00-6 a single category under TURA. Beginning with Potassium dichromate K Cr O 7778-50-9 reporting year 2012, hexavalent chromium 2 2 7 compounds are reportable under TURA as a Silver chromate Ag2CrO4 7784-01-2 separate category and are designated as a Higher Sodium chromate Na2CrO4 7775-11-3 Hazard Substance, which lowers the reporting Sodium dichromate 7789-12-0 threshold to 1,000 lb/year. -

20210311 IAEG AD-DSL V5.0 for Pdf.Xlsx
IAEGTM AD-DSL Release Version 4.1 12-30-2020 Authority: IAEG Identity: AD-DSL Version number: 4.1 Issue Date: 2020-12-30 Key Yellow shading indicates AD-DSL family group entries, which can be expanded to display a non-exhaustive list of secondary CAS numbers belonging to the family group Substance Identification Change Log IAEG Regulatory Date First Parent Group IAEG ID CAS EC Name Synonyms Revision Date ECHA ID Entry Type Criteria Added IAEG ID IAEG000001 1327-53-3 215-481-4 Diarsenic trioxide Arsenic trioxide R1;R2;D1 2015-03-17 2015-03-17 100.014.075 Substance Direct Entry IAEG000002 1303-28-2 215-116-9 Diarsenic pentaoxide Arsenic pentoxide; Arsenic oxide R1;R2;D1 2015-03-17 2015-03-17 100.013.743 Substance Direct Entry IAEG000003 15606-95-8 427-700-2 Triethyl arsenate R1;R2;D1 2015-03-17 2017-08-14 100.102.611 Substance Direct Entry IAEG000004 7778-39-4 231-901-9 Arsenic acid R1;R2;D1 2015-03-17 2015-03-17 100.029.001 Substance Direct Entry IAEG000005 3687-31-8 222-979-5 Trilead diarsenate R1;R2;D1 2015-03-17 2017-08-14 100.020.890 Substance Direct Entry IAEG000006 7778-44-1 231-904-5 Calcium arsenate R1;R2;D1 2015-03-17 2017-08-14 100.029.003 Substance Direct Entry IAEG000009 12006-15-4 234-484-1 Cadmium arsenide Tricadmium diarsenide R1;R2;D1 2017-08-14 2017-08-14 Substance Direct Entry IAEG000021 7440-41-7 231-150-7 Beryllium (Be) R2 2015-03-17 2019-01-24 Substance Direct Entry IAEG000022 1306-19-0 215-146-2 Cadmium oxide R1;R2;D1 2015-03-17 2017-08-14 100.013.770 Substance Direct Entry IAEG000023 10108-64-2 233-296-7 Cadmium -

CM300-011 Barium Chromate
Version: 1.0 Revision date: Safety Data Sheet 09/28/2015 Supersedes: 03/01/2013 Barium Chromate 1. PRODUCT AND COMPANY IDENTIFICATION 1.1. Product Identifiers Product Form: crystalline Substance Name: Barium Chromate CAS No.: 10294-40-3 Product Code: UIC, Inc. Catalog Number CM300-011 1.2. Intended Use of the Product Use of the substance/mixture: Laboratory chemicals, Manufacture of substances, Combustion tubes Name, Address, and Telephone of the Responsible Party UIC Inc 1225 Channahon Rd Joliet, IL 60436 Phone: (815) 744-4477 Fax: (815) 744-1561 Emergency Telephone Number For Chemical Emergency, Spill, Leak, Fire, Exposure, or Accident, call emergency number: 1-815-474-8753 2. Hazards Identification of the product 2.1. Classification of the substance or mixture Oxidizing solids (Category 2), H272 Acute toxicity, Oral (Category 4), H302 Acute toxicity, Inhalation (Category 4), H332 Carcinogenicity (Category 1A), H350 For the full text of the H-Statements mentioned in this Section, see Section 16. 2.2. GHS Label elements, including precautionary statements Pictogram Signal word Danger Hazard statement(s) H272 May intensify fire; oxidiser. H302 + H332 Harmful if swallowed or if inhaled H350 May cause cancer. Precautionary statement(s) P201 Obtain special instructions before use. P202 Do not handle until all safety precautions have been read and understood. P210 Keep away from heat. P220 Keep/Store away from clothing/ combustible materials. P221 Take any precaution to avoid mixing with combustibles. P261 Avoid breathing dust/ fume/ gas/ mist/ vapors/ spray. P264 Wash skin thoroughly after handling. P270 Do not eat, drink or smoke when using this product. -

Exploring Equilibria Name: Investigation 1: Chromate
Exploring Equilibria Name: ________________ Chem 112 This experiment explores a variety of equilibrium systems. A reference Table of Reactions is attached to aid in your explanations. In this qualitative lab, your observations, predictions, and explanations should be recorded in the spaces provided. Using what you know about equilibria and the Table of Reactions included, you should be able to explain all your observations in terms of the relative sizes of equilibrium constants and LeChatelier’s principle. Because your investigation is qualitative, the exact amounts of the solutions you work with are not critical. You will be transferring around 1 mL of solution, which is about the same as 20 drops. Don’t use much more than that of any solution. Notice and make use of the table of some common reaction found on the last page of the handout. Investigation 1: Chromate‐Dichromate Solutions 2‐ 2‐ There are two forms of chromium in high oxidation states: chromate ion (CrO4 ) and dichromate ion (Cr2O7 ). This experiment explores the equilibrium between them. What to do: 1. Place about 0.5 mL of 1 M potassium chromate solution in a small test tube. Describe the solution. Write a balanced equation for the dissolution of K2CrO4. 2. Add a few drops of 3 M sulfuric acid (H2SO4). Write your observations. 2‐ 2‐ 3. The chromate anion (CrO4 ) is converted into dichromate ion (Cr2O7 ) in the presence of acid. Write a net ionic equation for this reaction. 4. Add several drops of 6 M sodium hydroxide (NaOH). Record your observations and write a net ionic equation for what occurs. -

Substance Name: Strontium Chromate EC Number: 232-142-6 CAS Number: 7789-06-2
SVHC SUPPORT DOCUMENT – STRONTIUM CHROMATE Substance name: Strontium chromate EC number: 232-142-6 CAS number: 7789-06-2 MEMBER STATE COMMITTEE SUPPORT DOCUMENT FOR IDENTIFICATION OF STRONTIUM CHROMATE AS A SUBSTANCE OF VERY HIGH CONCERN BECAUSE OF ITS CMR PROPERTIES Adopted on 20 May 2011 1 SVHC SUPPORT DOCUMENT – STRONTIUM CHROMATE CONTENTS 1 IDENTITY OF THE SUBSTANCE AND PHYSICAL AND CHEMICAL PROPERTIES .................................4 1.1 Name and other identifiers of the substance ...................................................................................................4 1.2 Composition of the substance.........................................................................................................................5 1.3 Physico-chemical properties...........................................................................................................................6 2 HARMONISED CLASSIFICATION AND LABELLING....................................................................................7 3 ENVIRONMENTAL FATE PROPERTIES...........................................................................................................8 4 HUMAN HEALTH HAZARD ASSESSMENT.....................................................................................................8 5 ENVIRONMENTAL HAZARD ASSESSMENT .................................................................................................8 6 CONCLUSIONS ON THE SVHC PROPERTIES .................................................................................................8 -

Barium, Zinc and Strontium Yellows in Late 19Th–Early 20Th Century Oil Paintings Vanessa Otero1* , Marta F
Otero et al. Herit Sci (2017) 5:46 DOI 10.1186/s40494-017-0160-3 RESEARCH ARTICLE Open Access Barium, zinc and strontium yellows in late 19th–early 20th century oil paintings Vanessa Otero1* , Marta F. Campos1, Joana V. Pinto2, Márcia Vilarigues1, Leslie Carlyle1 and Maria João Melo1 Abstract This work focuses on the study of the 19th century yellow chromate pigments based on barium (BaCrO 4), zinc (4ZnCrO4 K2O 3H2O) and strontium (SrCrO4). These pigments, which are reported to shift in hue and darken, have been found· in· 19th century artworks. A better understanding of their historic manufacture will contribute to the visual/chemical interpretation of change in these colours. Research was carried out on the Winsor & Newton (W&N) 19th century archive database providing a unique insight into their manufacturing processes. One hundred and three production records were found, 69% for barium, 25% for zinc and 6% for strontium chromates, mainly under the names Lemon, Citron and Strontian Yellow, respectively. Analysis of the records shows that each pigment is charac- terised by only one synthetic pathway. The low number of records found for the production of strontium chromate suggests W&N was not selling this pigment formulation on a large scale. Furthermore, contrary to what the authors have discovered for W&N chrome yellow pigments, extenders were not added to these pigment formulations, most probably due to their lower tinting strength (TS). The latter was calculated in comparison to pure chrome yellow (PbCrO4, 100% TS) resulting in 92% for barium, 65% for zinc potassium and 78% for strontium chromate pigments. -
Safety Data Sheet According to 1907/2006/EC (REACH), 1272/2008/EC (CLP), and GHS
Page 1/15 Safety Data Sheet according to 1907/2006/EC (REACH), 1272/2008/EC (CLP), and GHS Printing date: September 10, 2014 Revision: September 10, 2014 SECTION 1: Identification of the substance/mixture and of the company/ undertaking · 1.1 Product identifier · Trade name: Stinger® 60-Caliber Rubber Balls · Article number: 1087 · 1.2 Relevant identified uses of the substance or mixture and uses advised against No further relevant information available. · Application of the substance / the mixture Explosive product. · 1.3 Details of the supplier of the Safety Data Sheet · Manufacturer/Supplier: Safariland, LLC 13386 International Parkway Jacksonville, FL 32218 Customer Care (800) 347-1200 · Further information obtainable from: Customer Care Department · 1.4 Emergency telephone number: ChemTel Inc. (800)255-3924, +1 (813)248-0585 SECTION 2: Hazards identification · 2.1 Classification of the substance or mixture · Classification according to Regulation (EC) No 1272/2008 The following Hazard Statements are applicable only to the EU regulations and not the US GHS regulation: H411. exploding bomb Expl. 1.4 H204 Fire or projection hazard. environment Aquat ic Chronic 2 H411 Toxic to aquatic life with long lasting effects. · Classification according to Directive 67/548/EEC or Directive 1999/45/EC N; Dangerous for the environment R51/53: Toxic to aquatic organisms, may cause long-term adverse effects in the aquatic environment. R5-44: Heating may cause an explosion. Risk of explosion if heated under confinement. · Information concerning particular hazards for human and environment: The product has to be labelled due to the calculation procedure of the "General Classification guideline for preparations of the EU" in the latest valid version. -

Screening Assessment for the Challenge C.I. Pigment Yellow 34
Screening Assessment for the Challenge C.I. Pigment Yellow 34 Chemical Abstracts Service Registry Number 1344-37-2 Environment Canada Health Canada November 2008 Screening Assessment CAS RN 1344-37-2 Synopsis Pursuant to section 74 of the Canadian Environmental Protection Act, 1999 (CEPA 1999), the Ministers of the Environment and of Health have conducted a screening assessment of C.I. Pigment Yellow 34, Chemical Abstracts Service Registry Number (CAS RN) 1344-37-2. The substance C.I. Pigment Yellow 34 was identified in the categorization of the Domestic Substances List as a high priority for action under the Ministerial Challenge. The substance was identified as a high priority because it was considered to pose greatest potential for exposure (GPE) to individuals in Canada and had been classified by other agencies on the basis of carcinogenicity, reproductive toxicity and developmental toxicity. The substance also met the ecological categorization criteria for persistence and inherent toxicity to aquatic organisms. Therefore, this assessment of C.I. Pigment Yellow 34 focuses on information relevant to the evaluation of both human health and ecological risks. In response to a notice issued under section 71 of CEPA 1999, in 2006 C.I. Pigment Yellow 34 was reported to be manufactured in and imported into Canada. After exports, the amount remaining for use in this country ranged between 1 000 000 and 10 000 000 kg. It is primarily used for plastic formulation for commercial applications and export; commercial, non-consumer paints and coatings; and commercial printing inks or coatings used for plastics and certain outdoor applications such as commercial identification decals. -

Developmental Process Study for Laboratory Scale Preparation of Barium Chromate
American-Eurasian Journal of Toxicological Sciences 6 (4): 114-119, 2014 ISSN 2079-2050 © IDOSI Publications, 2014 DOI: 10.5829/idosi.aejts.2014.6.4.1112 Developmental Process Study for Laboratory Scale Preparation of Barium Chromate 1Sheikh Asrar Ahmad, 1Maria Mustafa, 1Nafeesa Batool, 11Asad Gulzar, Sajid Mahmood Rao, 12Qamar Ali, Muhammad Nadeem Zafar, 3Adnan Akram, 4Rana Khalid Mahmood, 45Zia Ullah Khokhar and Muhammad Abdul Qadir 1Division of Science and Technology, University of Education, Lahore, Pakistan 2Department of Chemistry, University of the Gujrat, Pakistan 3PCSIR Laboratories complex, Feroz Pur Road Lahore, Pakistan 4Government Post graduate Islamia College Gujranwala, Pakistan 5Institute of Chemistry, University of the Punjab Lahore, Pakistan Abstract: Present work deals with the developmental process study for laboratory scale preparation of barium chromate. It is a cheaper and a viable method to prepare barium chromate which has very important industrial applications. Barium chromate was prepared by mixing different concentrations of sodium chromate and barium chloride. Percentage yields of barium chromate were determined 86%, 93%, 91%, 85% and 90% respectively. Different parameters such as temperature, pH, stirring speed, solubility were applied to get optimum yield. The optimum percentage yield was noted 93% for 0.3 M concentrations of sodium chromate and barium chloride at pH 7, 25°C without stirring. Barium was estimated gravimetrically in sample solution as barium sulfate and its optimum percentage yield was 46%. Percentage of chromium estimated by atomic adsorption spectrometry in sample solution was found to be 19.5%. The purity of barium chromate was checked by iodine titration method and maximum purity of sample solution was found to be 98%. -

Chemical Parameters Supporting Table
Regional Removal Management Level (RML) Chemical-specific Parameters Supporting Table May 2016 Contaminant Molecular Weight Volatility Parameters Melting Point Density Diffusivity in Air and Water Partition Coefficients Water Solubility Tapwater Dermal Parameters H` HLC Density Dia Diw Kd Koc log Kow S B τevent t* Kp 3 3 2 2 Analyte CAS No. MW MW Ref (unitless) (atm-m /mole) H` and HLC Ref VP VP Ref MP MP Ref (g/cm )Density Ref(cm /s) (cm /s) Dia and Diw Ref (L/kg) Kd Ref (L/kg) Koc Ref (unitless) log Kow Ref (mg/L) S Ref (unitless) (hr/event) (hr) (cm/hr) K Ref Acephate 30560-19-1 1.8E+02 PHYSPROP 2.0E-11 5.0E-13 EPI 1.7E-06 PHYSPROP 8.8E+01 PHYSPROP 1.4E+00 CRC89 3.7E-02 8.0E-06 WATER9 1.0E+01 EPI -8.5E-01 PHYSPROP 8.2E+05 PHYSPROP 2.1E-04 1.1E+00 2.7E+00 4.0E-05 EPI Acetaldehyde 75-07-0 4.4E+01 PHYSPROP 2.7E-03 6.7E-05 PHYSPROP 9.0E+02 PHYSPROP -1.2E+02 PHYSPROP 7.8E-01 CRC89 1.3E-01 1.4E-05 WATER9 1.0E+00 EPI -3.4E-01 PHYSPROP 1.0E+06 PHYSPROP 1.3E-03 1.9E-01 4.5E-01 5.3E-04 EPI Acetochlor 34256-82-1 2.7E+02 PHYSPROP 9.1E-07 2.2E-08 PHYSPROP 2.8E-05 PHYSPROP 1.1E+01 PubChem 1.1E+00 PubChem 2.2E-02 5.6E-06 WATER9 3.0E+02 EPI 3.0E+00 PHYSPROP 2.2E+02 PHYSPROP 3.1E-02 3.4E+00 8.2E+00 5.0E-03 EPI Acetone 67-64-1 5.8E+01 PHYSPROP 1.4E-03 3.5E-05 PHYSPROP 2.3E+02 PHYSPROP -9.5E+01 PHYSPROP 7.8E-01 CRC89 1.1E-01 1.2E-05 WATER9 2.4E+00 EPI -2.4E-01 PHYSPROP 1.0E+06 PHYSPROP 1.5E-03 2.2E-01 5.3E-01 5.1E-04 EPI Acetone Cyanohydrin 75-86-5 8.5E+01 PHYSPROP 8.1E-08 2.0E-09 PHYSPROP 3.4E-01 PHYSPROP -1.9E+01 PHYSPROP 9.3E-01 CRC89 8.6E-02 1.0E-05