Genome Rescue
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -
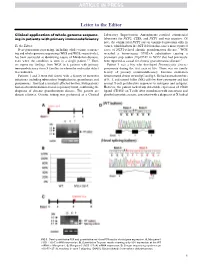
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -

KAT6A Syndrome: Genotype-Phenotype Correlation in 76 Patients with Pathogenic KAT6A Variants
KAT6A Syndrome: genotype-phenotype correlation in 76 patients with pathogenic KAT6A variants. Item Type Article Authors Kennedy, Joanna;Goudie, David;Blair, Edward;Chandler, Kate;Joss, Shelagh;McKay, Victoria;Green, Andrew;Armstrong, Ruth;Lees, Melissa;Kamien, Benjamin;Hopper, Bruce;Tan, Tiong Yang;Yap, Patrick;Stark, Zornitza;Okamoto, Nobuhiko;Miyake, Noriko;Matsumoto, Naomichi;Macnamara, Ellen;Murphy, Jennifer L;McCormick, Elizabeth;Hakonarson, Hakon;Falk, Marni J;Li, Dong;Blackburn, Patrick;Klee, Eric;Babovic- Vuksanovic, Dusica;Schelley, Susan;Hudgins, Louanne;Kant, Sarina;Isidor, Bertrand;Cogne, Benjamin;Bradbury, Kimberley;Williams, Mark;Patel, Chirag;Heussler, Helen;Duff- Farrier, Celia;Lakeman, Phillis;Scurr, Ingrid;Kini, Usha;Elting, Mariet;Reijnders, Margot;Schuurs-Hoeijmakers, Janneke;Wafik, Mohamed;Blomhoff, Anne;Ruivenkamp, Claudia A L;Nibbeling, Esther;Dingemans, Alexander J M;Douine, Emilie D;Nelson, Stanley F;Arboleda, Valerie A;Newbury-Ecob, Ruth DOI 10.1038/s41436-018-0259-2 Journal Genetics in medicine : official journal of the American College of Medical Genetics Download date 30/09/2021 21:53:13 Link to Item http://hdl.handle.net/10147/627191 Find this and similar works at - http://www.lenus.ie/hse HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Genet Med Manuscript Author . Author manuscript; Manuscript Author available in PMC 2019 October 01. Published in final edited form as: Genet Med. 2019 April ; 21(4): 850–860. doi:10.1038/s41436-018-0259-2. KAT6A Syndrome: Genotype-phenotype correlation in 76 patients with pathogenic KAT6A variants A full list of authors and affiliations appears at the end of the article. Abstract Purpose: Mutations in KAT6A have recently been identified as a cause of syndromic developmental delay. -

Thiadiazole Derivatives As Anticancer Agents
Pharmacological Reports (2020) 72:1079–1100 https://doi.org/10.1007/s43440-020-00154-7 REVIEW Thiadiazole derivatives as anticancer agents Monika Szeliga1 Received: 15 June 2020 / Revised: 13 August 2020 / Accepted: 20 August 2020 / Published online: 3 September 2020 © The Author(s) 2020 Abstract In spite of substantial progress made toward understanding cancer pathogenesis, this disease remains one of the leading causes of mortality. Thus, there is an urgent need to develop novel, more efective anticancer therapeutics. Thiadiazole ring is a versatile scafold widely studied in medicinal chemistry. Mesoionic character of this ring allows thiadiazole-containing compounds to cross cellular membrane and interact strongly with biological targets. Consequently, these compounds exert a broad spectrum of biological activities. This review presents the current state of knowledge on thiadiazole derivatives that demonstrate in vitro and/or in vivo efcacy across the cancer models with an emphasis on targets of action. The infuence of the substituent on the compounds’ activity is depicted. Furthermore, the results from clinical trials assessing thiadiazole- containing drugs in cancer patients are summarized. Keywords Thiadiazole derivatives · Cancer · Anticancer therapy · Clinical trials Introduction antiparasitic, anti-infammatory and anticancer activities [2]. Due to the mesoionic nature, thiadiazoles are able to According to the most recent data provided by the Interna- cross the cellular membranes. Their relatively good lipo- tional Agency for Research on Cancer (IARC), 18.1 mil- solubility is most likely attributed to the presence of the sul- lion new cases and 9.6 million cancer deaths were regis- phur atom [3]. The thiadiazole-containing drugs, including tered worldwide in 2018 [1]. -

Chr21 Protein-Protein Interactions: Enrichment in Products Involved in Intellectual Disabilities, Autism and Late Onset Alzheimer Disease
bioRxiv preprint doi: https://doi.org/10.1101/2019.12.11.872606; this version posted December 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Chr21 protein-protein interactions: enrichment in products involved in intellectual disabilities, autism and Late Onset Alzheimer Disease Julia Viard1,2*, Yann Loe-Mie1*, Rachel Daudin1, Malik Khelfaoui1, Christine Plancon2, Anne Boland2, Francisco Tejedor3, Richard L. Huganir4, Eunjoon Kim5, Makoto Kinoshita6, Guofa Liu7, Volker Haucke8, Thomas Moncion9, Eugene Yu10, Valérie Hindie9, Henri Bléhaut11, Clotilde Mircher12, Yann Herault13,14,15,16,17, Jean-François Deleuze2, Jean- Christophe Rain9, Michel Simonneau1, 18, 19, 20** and Aude-Marie Lepagnol- Bestel1** 1 Centre Psychiatrie & Neurosciences, INSERM U894, 75014 Paris, France 2 Laboratoire de génomique fonctionnelle, CNG, CEA, Evry 3 Instituto de Neurociencias CSIC-UMH, Universidad Miguel Hernandez-Campus de San Juan 03550 San Juan (Alicante), Spain 4 Department of Neuroscience, The Johns Hopkins University School of Medicine, Baltimore, MD 21205 USA 5 Center for Synaptic Brain Dysfunctions, Institute for Basic Science, Daejeon 34141, Republic of Korea 6 Department of Molecular Biology, Division of Biological Science, Nagoya University Graduate School of Science, Furo, Chikusa, Nagoya, Japan 7 Department of Biological Sciences, University of Toledo, Toledo, OH, 43606, USA 8 Leibniz Forschungsinstitut für Molekulare Pharmakologie -

ONCOPANEL (Popv3)
ONCOPANEL (POPv3) TEST INFORMATION BACKGROUND: Somatic genetic alterations in oncogenes and tumor-suppressor genes contribute to the pathogenesis and evolution of human cancers. These alterations can provide prognostic and predictive information and stratify cancers for targeted therapeutic information. We classify these alterations into five tiers using the following guidelines: Tier 1: The alteration has well-established published evidence confirming clinical utility in this tumor type, in at least one of the following contexts: predicting response to treatment with an FDA-approved therapy; assessing prognosis; establishing a definitive diagnosis; or conferring an inherited increased risk of cancer to this patient and family. Tier 2: The alteration may have clinical utility in at least one of the following contexts: selection of an investigational therapy in clinical trials for this cancer type; limited evidence of prognostic association; supportive of a specific diagnosis; proven association of response to treatment with an FDA-approved therapy in a different type of cancer; or similar to a different mutation with a proven association with response to treatment with an FDA-approved therapyin this type of cancer. Tier 3: The alteration is of uncertain clinical utility, but may have a role as suggested by at least one of the following: demonstration of association with response to treatment in this cancer type in preclinical studies (e.g., in vitro studies or animal models); alteration in a biochemical pathway that has other known, therapeutically-targetable alterations; alteration in a highly conserved region of the protein predicted, in silico, to alter protein function; or selection of an investigational therapy for a different cancer type. -

Neural Stem Cell-Derived Exosomes Revert HFD-Dependent Memory Impairment Via CREB-BDNF Signalling
International Journal of Molecular Sciences Article Neural Stem Cell-Derived Exosomes Revert HFD-Dependent Memory Impairment via CREB-BDNF Signalling Matteo Spinelli 1, Francesca Natale 1,2, Marco Rinaudo 1, Lucia Leone 1,2, Daniele Mezzogori 1, Salvatore Fusco 1,2,* and Claudio Grassi 1,2 1 Department of Neuroscience, Università Cattolica del Sacro Cuore, 00168 Rome, Italy; [email protected] (M.S.); [email protected] (F.N.); [email protected] (M.R.); [email protected] (L.L.); [email protected] (D.M.); [email protected] (C.G.) 2 Fondazione Policlinico Universitario Agostino Gemelli IRCCS, 00168 Rome, Italy * Correspondence: [email protected] Received: 6 October 2020; Accepted: 25 November 2020; Published: 26 November 2020 Abstract: Overnutrition and metabolic disorders impair cognitive functions through molecular mechanisms still poorly understood. In mice fed with a high fat diet (HFD) we analysed the expression of synaptic plasticity-related genes and the activation of cAMP response element-binding protein (CREB)-brain-derived neurotrophic factor (BDNF)-tropomyosin receptor kinase B (TrkB) signalling. We found that a HFD inhibited both CREB phosphorylation and the expression of a set of CREB target genes in the hippocampus. The intranasal administration of neural stem cell (NSC)-derived exosomes (exo-NSC) epigenetically restored the transcription of Bdnf, nNOS, Sirt1, Egr3, and RelA genes by inducing the recruitment of CREB on their regulatory sequences. Finally, exo-NSC administration rescued both BDNF signalling and memory in HFD mice. Collectively, our findings highlight novel mechanisms underlying HFD-related memory impairment and provide evidence of the potential therapeutic effect of exo-NSC against metabolic disease-related cognitive decline. -

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

Identification and Characterization of TPRKB Dependency in TP53 Deficient Cancers
Identification and Characterization of TPRKB Dependency in TP53 Deficient Cancers. by Kelly Kennaley A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Molecular and Cellular Pathology) in the University of Michigan 2019 Doctoral Committee: Associate Professor Zaneta Nikolovska-Coleska, Co-Chair Adjunct Associate Professor Scott A. Tomlins, Co-Chair Associate Professor Eric R. Fearon Associate Professor Alexey I. Nesvizhskii Kelly R. Kennaley [email protected] ORCID iD: 0000-0003-2439-9020 © Kelly R. Kennaley 2019 Acknowledgements I have immeasurable gratitude for the unwavering support and guidance I received throughout my dissertation. First and foremost, I would like to thank my thesis advisor and mentor Dr. Scott Tomlins for entrusting me with a challenging, interesting, and impactful project. He taught me how to drive a project forward through set-backs, ask the important questions, and always consider the impact of my work. I’m truly appreciative for his commitment to ensuring that I would get the most from my graduate education. I am also grateful to the many members of the Tomlins lab that made it the supportive, collaborative, and educational environment that it was. I would like to give special thanks to those I’ve worked closely with on this project, particularly Dr. Moloy Goswami for his mentorship, Lei Lucy Wang, Dr. Sumin Han, and undergraduate students Bhavneet Singh, Travis Weiss, and Myles Barlow. I am also grateful for the support of my thesis committee, Dr. Eric Fearon, Dr. Alexey Nesvizhskii, and my co-mentor Dr. Zaneta Nikolovska-Coleska, who have offered guidance and critical evaluation since project inception. -

(12) United States Patent (10) Patent No.: US 8,323,640 B2 Sakuraba Et Al
USOO8323640B2 (12) United States Patent (10) Patent No.: US 8,323,640 B2 Sakuraba et al. (45) Date of Patent: *Dec. 4, 2012 (54) HIGHLY FUNCTIONAL ENZYME HAVING OTHER PUBLICATIONS O-GALACTOSIDASE ACTIVITY Broun et al., Catalytic plasticity of fatty acid modification enzymes (75) Inventors: Hitoshi Sakuraba, Abiko (JP); Youichi underlying chemical diversity of plant lipids. Science, 1998, vol. 282: Tajima, Tokyo (JP); Mai Ito, Tokyo 1315-1317. (JP); Seiichi Aikawa, Tokyo (JP); Cameron ER. Recent advances in transgenic technology. 1997, vol. T: 253-265. Fumiko Aikawa, Tokyo (JP) Chica et al., Semi-rational approaches to engineering enzyme activ (73) Assignees: Tokyo Metropolitan Organization For ity: combining the benefits of directed evolution and rational design. Curr, opi. Biotechnol., 2005, vol. 16:378-384. Medical Research, Tokyo (JP); Altif Couzin et al. As Gelsinger case ends, Gene therapy Suffers another Laboratories, Tokyo (JP) blow. Science, 2005, vol. 307: 1028. Devos et al., Practical Limits of Function prediction. Proteins: Struc (*) Notice: Subject to any disclaimer, the term of this ture, Function, and Genetics. 2000, vol. 41: 98-107. patent is extended or adjusted under 35 Donsante et al., AAV vector integration sites in mouse hapatocellular U.S.C. 154(b) by 0 days. carcinoma. Science, 2007. vol. 317: 477. Juengst ET., What next for human gene therapy? BMJ., 2003, vol. This patent is Subject to a terminal dis 326: 1410-1411. claimer. Kappel et al., Regulating gene expression in transgenic animals. Current Opinion in Biotechnology 1992, vol. 3: 548-553. (21) Appl. No.: 13/052,632 Kimmelman J. Recent developments in gene transfer: risk and eth ics. -

Supporting Information
Supporting Information Figure S1. The functionality of the tagged Arp6 and Swr1 was confirmed by monitoring cell growth and sensitivity to hydeoxyurea (HU). Five-fold serial dilutions of each strain were plated on YPD with or without 50 mM HU and incubated at 30°C or 37°C for 3 days. Figure S2. Localization of Arp6 and Swr1 on chromosome 3. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position of Tel 3L, Tel 3R, CEN3, and the RP gene are shown under the panels. Figure S3. Localization of Arp6 and Swr1 on chromosome 4. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) in the whole chromosome region are compared. The position of Tel 4L, Tel 4R, CEN4, SWR1, and RP genes are shown under the panels. Figure S4. Localization of Arp6 and Swr1 on the region including the SWR1 gene of chromosome 4. The binding of Arp6- FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position and orientation of the SWR1 gene is shown. Figure S5. Localization of Arp6 and Swr1 on chromosome 5. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position of Tel 5L, Tel 5R, CEN5, and the RP genes are shown under the panels. Figure S6. Preferential localization of Arp6 and Swr1 in the 5′ end of genes. Vertical bars represent the binding ratio of proteins in each locus. -

Supplementary Information Biocementation of Soil by Calcite/Aragonite Precipitation Using Pseudomonas Azotoformans and Citrobact
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2019 Supplementary Information Biocementation of soil by calcite/aragonite precipitation using Pseudomonas azotoformans and Citrobacter freundii derived enzymes Heba Abdel-Aleem1, Tarek Dishisha1, Amal Saafan2, Abduallah A. AbouKhadra3 and Yasser Gaber1,4,* 1Department of Microbiology and Immunology, Faculty of Pharmacy, Beni-Suef University, 62511, Beni-Suef, Egypt 2Department of Pharmaceutical Microbiology, Faculty of Pharmacy, Menoufia university, Shebeen El-kom, Egypt 3Department of Civil Engineering, Facutly of Engineering, Beni-Suef University, 62511, Beni- Suef, Egypt 4Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Mutah University, Al-karak, 61710, Jordan *Corresponding author: E-mail: [email protected] Tel. int +20 82 216 2133 ; Fax. int +20 82 216 2133 1 Supplementary Table 1: strains identified using BCL card (Gram positive spore forming bacilli) Sample/test A1 B1 B2 Y1a Y2a BXYL - - - - - BXYL (betaxylosidase), LysA - - - - - LysA (L- lysine arylamidase), AspA - - - + - AspA (L aspartate arylamidase), LeuA + + + + + LeuA (leucine arylamidase), PheA - + - - - PheA (phenylalanine arylamidase), ProA - - - + - ProA (L-proline arylamidase), BGAL - - - - - BGAL (beta galactosidase), PyrA + + + + + PyrA (L-pyrrolydonyl arylamidase), AGAL - - - - - AGAL (alpha galactosidase), AlaA - - - + - AlaA (alanine arylamidase), TyrA - - - - - TyrA (tyrosine arylamidase), BNAG + + + + + BNAG (beta-N-acetyl