Moran's Autocorrelation Coefficient in Comparative Methods
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
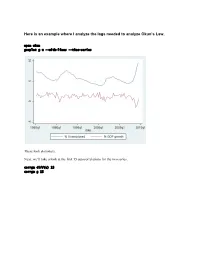
Here Is an Example Where I Analyze the Lags Needed to Analyze Okun's
Here is an example where I analyze the lags needed to analyze Okun’s Law. open okun gnuplot g u --with-lines --time-series These look stationary. Next, we’ll take a look at the first 15 autocorrelations for the two series. corrgm diff(u) 15 corrgm g 15 These are autocorrelated and so are these. I would expect that a simple regression will not be sufficient to model this relationship. Let’s try anyway. Run a simple regression and look at the residuals ols diff(u) const g series ehat=$uhat gnuplot ehat --with-lines --time-series These look positively autocorrelated to me. Notice how the residuals fall below zero for a while, then rise above for a while, and repeat this pattern. Take a look at the correlogram. The first autocorrelation is significant. More lags of D.u or g will probably fix this. LM test of residuals smpl 1985.2 2009.3 ols diff(u) const g modeltab add modtest 1 --autocorr --quiet modtest 2 --autocorr --quiet modtest 3 --autocorr --quiet modtest 4 --autocorr --quiet Breusch-Godfrey test for first-order autocorrelation Alternative statistic: TR^2 = 6.576105, with p-value = P(Chi-square(1) > 6.5761) = 0.0103 Breusch-Godfrey test for autocorrelation up to order 2 Alternative statistic: TR^2 = 7.922218, with p-value = P(Chi-square(2) > 7.92222) = 0.019 Breusch-Godfrey test for autocorrelation up to order 3 Alternative statistic: TR^2 = 11.200978, with p-value = P(Chi-square(3) > 11.201) = 0.0107 Breusch-Godfrey test for autocorrelation up to order 4 Alternative statistic: TR^2 = 11.220956, with p-value = P(Chi-square(4) > 11.221) = 0.0242 Yep, that’s not too good. -

Alternative Tests for Time Series Dependence Based on Autocorrelation Coefficients
Alternative Tests for Time Series Dependence Based on Autocorrelation Coefficients Richard M. Levich and Rosario C. Rizzo * Current Draft: December 1998 Abstract: When autocorrelation is small, existing statistical techniques may not be powerful enough to reject the hypothesis that a series is free of autocorrelation. We propose two new and simple statistical tests (RHO and PHI) based on the unweighted sum of autocorrelation and partial autocorrelation coefficients. We analyze a set of simulated data to show the higher power of RHO and PHI in comparison to conventional tests for autocorrelation, especially in the presence of small but persistent autocorrelation. We show an application of our tests to data on currency futures to demonstrate their practical use. Finally, we indicate how our methodology could be used for a new class of time series models (the Generalized Autoregressive, or GAR models) that take into account the presence of small but persistent autocorrelation. _______________________________________________________________ An earlier version of this paper was presented at the Symposium on Global Integration and Competition, sponsored by the Center for Japan-U.S. Business and Economic Studies, Stern School of Business, New York University, March 27-28, 1997. We thank Paul Samuelson and the other participants at that conference for useful comments. * Stern School of Business, New York University and Research Department, Bank of Italy, respectively. 1 1. Introduction Economic time series are often characterized by positive -

3 Autocorrelation
3 Autocorrelation Autocorrelation refers to the correlation of a time series with its own past and future values. Autocorrelation is also sometimes called “lagged correlation” or “serial correlation”, which refers to the correlation between members of a series of numbers arranged in time. Positive autocorrelation might be considered a specific form of “persistence”, a tendency for a system to remain in the same state from one observation to the next. For example, the likelihood of tomorrow being rainy is greater if today is rainy than if today is dry. Geophysical time series are frequently autocorrelated because of inertia or carryover processes in the physical system. For example, the slowly evolving and moving low pressure systems in the atmosphere might impart persistence to daily rainfall. Or the slow drainage of groundwater reserves might impart correlation to successive annual flows of a river. Or stored photosynthates might impart correlation to successive annual values of tree-ring indices. Autocorrelation complicates the application of statistical tests by reducing the effective sample size. Autocorrelation can also complicate the identification of significant covariance or correlation between time series (e.g., precipitation with a tree-ring series). Autocorrelation implies that a time series is predictable, probabilistically, as future values are correlated with current and past values. Three tools for assessing the autocorrelation of a time series are (1) the time series plot, (2) the lagged scatterplot, and (3) the autocorrelation function. 3.1 Time series plot Positively autocorrelated series are sometimes referred to as persistent because positive departures from the mean tend to be followed by positive depatures from the mean, and negative departures from the mean tend to be followed by negative departures (Figure 3.1). -

Statistical Tool for Soil Biology : 11. Autocorrelogram and Mantel Test
‘i Pl w f ’* Em J. 1996, (4), i Soil BioZ., 32 195-203 Statistical tool for soil biology. XI. Autocorrelogram and Mantel test Jean-Pierre Rossi Laboratoire d Ecologie des Sols Trofiicaux, ORSTOM/Université Paris 6, 32, uv. Varagnat, 93143 Bondy Cedex, France. E-mail: rossij@[email protected]. fr. Received October 23, 1996; accepted March 24, 1997. Abstract Autocorrelation analysis by Moran’s I and the Geary’s c coefficients is described and illustrated by the analysis of the spatial pattern of the tropical earthworm Clzuniodrilus ziehe (Eudrilidae). Simple and partial Mantel tests are presented and illustrated through the analysis various data sets. The interest of these methods for soil ecology is discussed. Keywords: Autocorrelation, correlogram, Mantel test, geostatistics, spatial distribution, earthworm, plant-parasitic nematode. L’outil statistique en biologie du sol. X.?. Autocorrélogramme et test de Mantel. Résumé L’analyse de l’autocorrélation par les indices I de Moran et c de Geary est décrite et illustrée à travers l’analyse de la distribution spatiale du ver de terre tropical Chuniodi-ibs zielae (Eudrilidae). Les tests simple et partiel de Mantel sont présentés et illustrés à travers l’analyse de jeux de données divers. L’intérêt de ces méthodes en écologie du sol est discuté. Mots-clés : Autocorrélation, corrélogramme, test de Mantel, géostatistiques, distribution spatiale, vers de terre, nématodes phytoparasites. INTRODUCTION the variance that appear to be related by a simple power law. The obvious interest of that method is Spatial heterogeneity is an inherent feature of that samples from various sites can be included in soil faunal communities with significant functional the analysis. -

Applied Time Series Analysis
Applied Time Series Analysis SS 2018 February 12, 2018 Dr. Marcel Dettling Institute for Data Analysis and Process Design Zurich University of Applied Sciences CH-8401 Winterthur Table of Contents 1 INTRODUCTION 1 1.1 PURPOSE 1 1.2 EXAMPLES 2 1.3 GOALS IN TIME SERIES ANALYSIS 8 2 MATHEMATICAL CONCEPTS 11 2.1 DEFINITION OF A TIME SERIES 11 2.2 STATIONARITY 11 2.3 TESTING STATIONARITY 13 3 TIME SERIES IN R 15 3.1 TIME SERIES CLASSES 15 3.2 DATES AND TIMES IN R 17 3.3 DATA IMPORT 21 4 DESCRIPTIVE ANALYSIS 23 4.1 VISUALIZATION 23 4.2 TRANSFORMATIONS 26 4.3 DECOMPOSITION 29 4.4 AUTOCORRELATION 50 4.5 PARTIAL AUTOCORRELATION 66 5 STATIONARY TIME SERIES MODELS 69 5.1 WHITE NOISE 69 5.2 ESTIMATING THE CONDITIONAL MEAN 70 5.3 AUTOREGRESSIVE MODELS 71 5.4 MOVING AVERAGE MODELS 85 5.5 ARMA(P,Q) MODELS 93 6 SARIMA AND GARCH MODELS 99 6.1 ARIMA MODELS 99 6.2 SARIMA MODELS 105 6.3 ARCH/GARCH MODELS 109 7 TIME SERIES REGRESSION 113 7.1 WHAT IS THE PROBLEM? 113 7.2 FINDING CORRELATED ERRORS 117 7.3 COCHRANE‐ORCUTT METHOD 124 7.4 GENERALIZED LEAST SQUARES 125 7.5 MISSING PREDICTOR VARIABLES 131 8 FORECASTING 137 8.1 STATIONARY TIME SERIES 138 8.2 SERIES WITH TREND AND SEASON 145 8.3 EXPONENTIAL SMOOTHING 152 9 MULTIVARIATE TIME SERIES ANALYSIS 161 9.1 PRACTICAL EXAMPLE 161 9.2 CROSS CORRELATION 165 9.3 PREWHITENING 168 9.4 TRANSFER FUNCTION MODELS 170 10 SPECTRAL ANALYSIS 175 10.1 DECOMPOSING IN THE FREQUENCY DOMAIN 175 10.2 THE SPECTRUM 179 10.3 REAL WORLD EXAMPLE 186 11 STATE SPACE MODELS 187 11.1 STATE SPACE FORMULATION 187 11.2 AR PROCESSES WITH MEASUREMENT NOISE 188 11.3 DYNAMIC LINEAR MODELS 191 ATSA 1 Introduction 1 Introduction 1.1 Purpose Time series data, i.e. -

Quantitative Risk Management in R
QUANTITATIVE RISK MANAGEMENT IN R Characteristics of volatile return series Quantitative Risk Management in R Log-returns compared with iid data ● Can financial returns be modeled as independent and identically distributed (iid)? ● Random walk model for log asset prices ● Implies that future price behavior cannot be predicted ● Instructive to compare real returns with iid data ● Real returns o!en show volatility clustering QUANTITATIVE RISK MANAGEMENT IN R Let’s practice! QUANTITATIVE RISK MANAGEMENT IN R Estimating serial correlation Quantitative Risk Management in R Sample autocorrelations ● Sample autocorrelation function (acf) measures correlation between variables separated by lag (k) ● Stationarity is implicitly assumed: ● Expected return constant over time ● Variance of return distribution always the same ● Correlation between returns k apart always the same ● Notation for sample autocorrelation: ⇢ˆ(k) Quantitative Risk Management in R The sample acf plot or correlogram > acf(ftse) Quantitative Risk Management in R The sample acf plot or correlogram > acf(abs(ftse)) QUANTITATIVE RISK MANAGEMENT IN R Let’s practice! QUANTITATIVE RISK MANAGEMENT IN R The Ljung-Box test Quantitative Risk Management in R Testing the iid hypothesis with the Ljung-Box test ● Numerical test calculated from squared sample autocorrelations up to certain lag ● Compared with chi-squared distribution with k degrees of freedom (df) ● Should also be carried out on absolute returns k ⇢ˆ(j)2 X2 = n(n + 2) n j j=1 X − Quantitative Risk Management in R Example of -

Autocorrelation and Seasonality Otexts.Com/Fpp/2/ Otexts.Com/Fpp/6/1
Rob J Hyndman Forecasting using 3. Autocorrelation and seasonality OTexts.com/fpp/2/ OTexts.com/fpp/6/1 Forecasting using R 1 Outline 1 Time series graphics 2 Seasonal or cyclic? 3 Autocorrelation Forecasting using R Time series graphics 2 Time series graphics Time plots R command: plot or plot.ts Seasonal plots R command: seasonplot Seasonal subseries plots R command: monthplot Lag plots R command: lag.plot ACF plots R command: Acf Forecasting using R Time series graphics 3 Time series graphics Economy class passengers: Melbourne−Sydney plot(melsyd[,"Economy.Class"]) 30 25 20 15 Thousands 10 5 0 1988 1989 1990 1991 1992 1993 Year Forecasting using R Time series graphics 4 Time series graphics Antidiabetic drug sales 30 > plot(a10) 25 20 15 $ million 10 5 1995 2000 2005 Year Forecasting using R Time series graphics 5 Time series graphics Seasonal plot: antidiabetic drug sales 30 2008 ● 2007 ● ● 2007 ● 25 ● ● 2006 ● ● ● ● ● ● 2006 ● ● ● ● 2005 ● ● ● ● 2005 20 ● ● ● ● 2004 ● ● 2004 ● ● ● ● ● ● ● ● ● 2003 ● ● ● ● ● ● 2003 2002 ● ● ● ● ● ● $ million ● 2002 15 ● ● 2001 ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● 2001 2000 ● ● ● ● ● ● ● 2000 ● ● ● 1999 ● ● ● ● ● ● ● ● ● ● 1998 1999 ● ● ● ● ● ● ● 1997 10 ● ● ● ● ● ● ● ● ● ● ● 1998 ● ● 1996 19961997 ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● 19931995 ● ● ● ● ● 19941995 ● ● ● ● ● ● ● ● ● ● ● ● 1993 ● ● ● 1994 ● ● ● ● ● ● ● 1992 ● ● ● ● ● ● ● ● ● 5 1992 ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● 1991 ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● Jan Feb Mar Apr May Jun Jul Aug Sep Oct Nov Dec Year Forecasting using R Time series graphics 6 Seasonal plots Data plotted against the individual “seasons” in which the data were observed. (In this case a “season” is a month.) Something like a time plot except that the data from each season are overlapped. Enables the underlying seasonal pattern to be seen more clearly, and also allows any substantial departures from the seasonal pattern to be easily identified. -

Chapter 5: Spatial Autocorrelation Statistics
Chapter 5: Spatial Autocorrelation Statistics Ned Levine1 Ned Levine & Associates Houston, TX 1 The author would like to thank Dr. David Wong for help with the Getis-Ord >G= and local Getis-Ord statistics. Table of Contents Spatial Autocorrelation 5.1 Spatial Autocorrelation Statistics for Zonal Data 5.3 Indices of Spatial Autocorrelation 5.3 Assigning Point Data to Zones 5.3 Spatial Autocorrelation Statistics for Attribute Data 5.5 Moran=s “I” Statistic 5.5 Adjust for Small Distances 5.6 Testing the Significance of Moran’s “I” 5.7 Example: Testing Houston Burglaries with Moran’s “I” 5.8 Comparing Moran’s “I” for Two Distributions 5.10 Geary=s “C” Statistic 5.10 Adjusted “C” 5.13 Adjust for Small Distances 5.13 Testing the Significance of Geary’s “C” 5.14 Example: Testing Houston Burglaries with Geary’s “C” 5.14 Getis-Ord “G” Statistic 5.16 Testing the Significance of “G” 5.18 Simulating Confidence Intervals for “G” 5.20 Example: Testing Simulated Data with the Getis-Ord AG@ 5.20 Example: Testing Houston Burglaries with the Getis-Ord AG@ 5.25 Use and Limitations of the Getis-Ord AG@ 5.25 Moran Correlogram 5.26 Adjust for Small Distances 5.27 Simulation of Confidence Intervals 5.27 Example: Moran Correlogram of Baltimore County Vehicle Theft and Population 5.27 Uses and Limitations of the Moran Correlogram 5.30 Geary Correlogram 5.34 Adjust for Small Distances 5.34 Geary Correlogram Simulation of Confidence Intervals 5.34 Example: Geary Correlogram of Baltimore County Vehicle Thefts 5.34 Uses and Limitations of the Geary Correlogram 5.35 Table of Contents (continued) Getis-Ord Correlogram 5.35 Getis-Ord Simulation of Confidence Intervals 5.37 Example: Getis-Ord Correlogram of Baltimore County Vehicle Thefts 5.37 Uses and Limitations of the Getis-Ord Correlogram 5.39 Running the Spatial Autocorrelation Routines 5.39 Guidelines for Examining Spatial Autocorrelation 5.39 References 5.42 Attachments 5.44 A. -

Correlation and Regression Analysis
OIC ACCREDITATION CERTIFICATION PROGRAMME FOR OFFICIAL STATISTICS Correlation and Regression Analysis TEXTBOOK ORGANISATION OF ISLAMIC COOPERATION STATISTICAL ECONOMIC AND SOCIAL RESEARCH AND TRAINING CENTRE FOR ISLAMIC COUNTRIES OIC ACCREDITATION CERTIFICATION PROGRAMME FOR OFFICIAL STATISTICS Correlation and Regression Analysis TEXTBOOK {{Dr. Mohamed Ahmed Zaid}} ORGANISATION OF ISLAMIC COOPERATION STATISTICAL ECONOMIC AND SOCIAL RESEARCH AND TRAINING CENTRE FOR ISLAMIC COUNTRIES © 2015 The Statistical, Economic and Social Research and Training Centre for Islamic Countries (SESRIC) Kudüs Cad. No: 9, Diplomatik Site, 06450 Oran, Ankara – Turkey Telephone +90 – 312 – 468 6172 Internet www.sesric.org E-mail [email protected] The material presented in this publication is copyrighted. The authors give the permission to view, copy download, and print the material presented that these materials are not going to be reused, on whatsoever condition, for commercial purposes. For permission to reproduce or reprint any part of this publication, please send a request with complete information to the Publication Department of SESRIC. All queries on rights and licenses should be addressed to the Statistics Department, SESRIC, at the aforementioned address. DISCLAIMER: Any views or opinions presented in this document are solely those of the author(s) and do not reflect the views of SESRIC. ISBN: xxx-xxx-xxxx-xx-x Cover design by Publication Department, SESRIC. For additional information, contact Statistics Department, SESRIC. i CONTENTS Acronyms -

Reference Manual
PAST PAleontological STatistics Version 3.16 Reference manual Øyvind Hammer Natural History Museum University of Oslo [email protected] 1999-2017 1 Contents Welcome to the PAST! ......................................................................................................................... 11 Installation ........................................................................................................................................... 12 Quick start ............................................................................................................................................ 13 How do I export graphics? ................................................................................................................ 13 How do I organize data into groups? ................................................................................................ 13 The spreadsheet and the Edit menu .................................................................................................... 14 Entering data .................................................................................................................................... 14 Selecting areas ................................................................................................................................. 14 Moving a row or a column................................................................................................................ 14 Renaming rows and columns .......................................................................................................... -

Forecasting and Predictive Analytics with Forecast X, 7E (Keating) Chapter 2 the Forecast Process, Data Considerations, and Model Selection
Forecasting and Predictive Analytics with Forecast X, 7e (Keating) Chapter 2 The Forecast Process, Data Considerations, and Model Selection 1) Why are forecasting textbooks full of applied statistics? A) Statistics is the study of uncertainty. B) Real-world business decisions involve risk and uncertainty. C) Forecasting attempts to generate certainty out of uncertain events. D) Forecasting ultimately deals with probability. E) All of the options are correct. 2) Which of the following is not part of the recommended nine-step forecast process? A) What role do forecasts play in the business decision process? B) What exactly is to be forecast? C) How urgent is the forecast? D) Is there enough data? E) All of the options are correct. 3) Of the following model selection criteria, which is often the most important in determining the appropriate forecast method? A) Technical background of the forecast user B) Patterns the data have exhibited in the past C) How much money is in the forecast budget? D) What is the forecast horizon? E) When is the forecast needed? 4) Time series data of a typical The GAP store should show which of the following data patterns? A) Trend B) Seasonal C) Cyclical D) Random E) All of the options are correct. 5) Which of the following is incorrect? A) The forecaster should be able to defend why a particular model or procedure has been chosen. B) Forecast errors should be discussed in an objective manner to maximize management's confidence in the forecast process. C) Forecast errors should not be discussed since most people know that forecasting is an inexact science. -

STAT 153: Introduction to Time Series
STAT 153: Introduction to Time Series Instructor: Aditya Guntuboyina Lectures: 12:30 pm - 2 pm (Tuesdays and Thursdays) Office Hours: 10 am - 11 am (Tuesdays and Thursdays) 423 Evans Hall GSI: Brianna Heggeseth Section: 10 am - 12 pm or 12 pm - 2 pm (Fridays) Office Hours and Location: TBA Announcements, Lecture slides, Assignments, etc. will be posted on the course site at bspace. Tuesday, January 17, 12 Course Outline A Time Series is a set of numerical observations, each one being recorded at a specific time. Examples of Time Series data are ubiquitous. The aim of this course is to teach you how to analyze such data. Tuesday, January 17, 12 Population of the United States 3.0e+08 2.0e+08 US Population 1.0e+08 0.0e+00 1800 1850 1900 1950 2000 Year(once every ten years) Tuesday, January 17, 12 Course Outline (continued) There are two approaches to time series analysis: • Time Domain Approach • Frequency Domain Approach (also known as the Spectral or Fourier analysis of time series) Very roughly, 60% of the course will be on Time Domain methods and 40% on Frequency Domain methods. Tuesday, January 17, 12 Time Domain Approach Seeks an answer to the following question: Given the observed time series, how does one guess future values? Forecasting or Prediction Tuesday, January 17, 12 Time Domain (continued) Forecasting is carried out through three steps: • Find a MODEL that adequately describes the evolution of the observed time series over time. • Fit this model to the data. In other words, estimate the parameters of the model.