Molecular and Functional Basis of a Novel Amazonian Dark Earth Esterase 1 (Ade1) with Hysteresis Behavior and Quorum-Quenching Activity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MIAMI UNIVERSITY the Graduate School Certificate for Approving The
MIAMI UNIVERSITY The Graduate School Certificate for Approving the Dissertation We hereby approve the Dissertation of Matthew F. Rouhier Candidate for the Degree: Doctor of Philosophy ___________________________________________________________ Director (Dr. Ann Hagerman) ___________________________________________________________ Reader (Dr.Chris Makaroff) ___________________________________________________________ Reader (Dr.Gary Lorigan) __________________________________________________________ Reader (Dr. Richard Taylor) _________________________________________________________ Graduate School Representative (Dr. Paul James) ! ABSTRACT CHARACTERIZATION OF YDR036C FROM Saccharomyces cerevisiae by Matthew F. Rouhier Beta-hydroxyisobutyryl-CoA (HIBYL-CoA) hydrolases are found ubiquitously in eukaryotes where they function in the catabolism of valine. A homologous enzyme (YDR036C) is also present in yeast where valine catabolism is distinctly different and does not require a HIBYL-CoA hydrolase. Like the other eukaryotic hydrolases, the yeast hydrolase is a member of the crotonase super-family which catalyzes various reactions using CoA thioester substrates. Crotonases typically function in fatty acid degradation which is peroxisomal in yeast while YDR036C is found strictly in mitochondria. Like other HIBYL-CoA hydrolases the yeast enzyme demonstrated activity toward beta-hydroxyacyl-CoAs, but unlike the other HIBYL-CoA hydrolases the yeast activity is greatest with beta-hydroxypropionyl-CoA (3-HP-CoA). Further characterization of the active site has determined that the preference of the hydrolases for 3-HP- CoA or HIBYL-CoA is dependent upon the residue at position 177. The glutamate at position 121 is responsible for the coordination with the beta-hydroxyl group and replacement with valine renders the enzyme non-specific for a hydroxyl group. Another unique feature to yeast hydrolases is the presence of a C-terminal tail of 80 amino acids, which when removed renders the enzyme inactive, suggesting that it may play a role in substrate binding. -

Type of the Paper (Article
Supplementary Material A Proteomics Study on the Mechanism of Nutmeg-induced Hepatotoxicity Wei Xia 1, †, Zhipeng Cao 1, †, Xiaoyu Zhang 1 and Lina Gao 1,* 1 School of Forensic Medicine, China Medical University, Shenyang 110122, P. R. China; lessen- [email protected] (W.X.); [email protected] (Z.C.); [email protected] (X.Z.) † The authors contributed equally to this work. * Correspondence: [email protected] Figure S1. Table S1. Peptide fraction separation liquid chromatography elution gradient table. Time (min) Flow rate (mL/min) Mobile phase A (%) Mobile phase B (%) 0 1 97 3 10 1 95 5 30 1 80 20 48 1 60 40 50 1 50 50 53 1 30 70 54 1 0 100 1 Table 2. Liquid chromatography elution gradient table. Time (min) Flow rate (nL/min) Mobile phase A (%) Mobile phase B (%) 0 600 94 6 2 600 83 17 82 600 60 40 84 600 50 50 85 600 45 55 90 600 0 100 Table S3. The analysis parameter of Proteome Discoverer 2.2. Item Value Type of Quantification Reporter Quantification (TMT) Enzyme Trypsin Max.Missed Cleavage Sites 2 Precursor Mass Tolerance 10 ppm Fragment Mass Tolerance 0.02 Da Dynamic Modification Oxidation/+15.995 Da (M) and TMT /+229.163 Da (K,Y) N-Terminal Modification Acetyl/+42.011 Da (N-Terminal) and TMT /+229.163 Da (N-Terminal) Static Modification Carbamidomethyl/+57.021 Da (C) 2 Table S4. The DEPs between the low-dose group and the control group. Protein Gene Fold Change P value Trend mRNA H2-K1 0.380 0.010 down Glutamine synthetase 0.426 0.022 down Annexin Anxa6 0.447 0.032 down mRNA H2-D1 0.467 0.002 down Ribokinase Rbks 0.487 0.000 -

(10) Patent No.: US 8119385 B2
US008119385B2 (12) United States Patent (10) Patent No.: US 8,119,385 B2 Mathur et al. (45) Date of Patent: Feb. 21, 2012 (54) NUCLEICACIDS AND PROTEINS AND (52) U.S. Cl. ........................................ 435/212:530/350 METHODS FOR MAKING AND USING THEMI (58) Field of Classification Search ........................ None (75) Inventors: Eric J. Mathur, San Diego, CA (US); See application file for complete search history. Cathy Chang, San Diego, CA (US) (56) References Cited (73) Assignee: BP Corporation North America Inc., Houston, TX (US) OTHER PUBLICATIONS c Mount, Bioinformatics, Cold Spring Harbor Press, Cold Spring Har (*) Notice: Subject to any disclaimer, the term of this bor New York, 2001, pp. 382-393.* patent is extended or adjusted under 35 Spencer et al., “Whole-Genome Sequence Variation among Multiple U.S.C. 154(b) by 689 days. Isolates of Pseudomonas aeruginosa” J. Bacteriol. (2003) 185: 1316 1325. (21) Appl. No.: 11/817,403 Database Sequence GenBank Accession No. BZ569932 Dec. 17. 1-1. 2002. (22) PCT Fled: Mar. 3, 2006 Omiecinski et al., “Epoxide Hydrolase-Polymorphism and role in (86). PCT No.: PCT/US2OO6/OOT642 toxicology” Toxicol. Lett. (2000) 1.12: 365-370. S371 (c)(1), * cited by examiner (2), (4) Date: May 7, 2008 Primary Examiner — James Martinell (87) PCT Pub. No.: WO2006/096527 (74) Attorney, Agent, or Firm — Kalim S. Fuzail PCT Pub. Date: Sep. 14, 2006 (57) ABSTRACT (65) Prior Publication Data The invention provides polypeptides, including enzymes, structural proteins and binding proteins, polynucleotides US 201O/OO11456A1 Jan. 14, 2010 encoding these polypeptides, and methods of making and using these polynucleotides and polypeptides. -

Isomer-Specific Comparisons of the Hydrolysis of Synthetic Pyrethroids and Their Fluorogenic Analogues by Esterases from The
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Pesticide Biochemistry and Physiology 121 (2015) 102–106 Contents lists available at ScienceDirect Pesticide Biochemistry and Physiology journal homepage: www.elsevier.com/locate/pest Isomer-specific comparisons of the hydrolysis of synthetic pyrethroids and their fluorogenic analogues by esterases from the cotton bollworm Helicoverpa armigera G. Yuan a,b, Y. Li c, C.A. Farnsworth d,e,f, C.W. Coppin d, A.L. Devonshire d, C. Scott d, R.J. Russell d, Y. Wu b, J.G. Oakeshott d,* a Key laboratory of Entomology and Pest Control Engineering, College of Plant Protection, Southwest University, Chongqing 400716, China b Department of Entomology, College of Plant Protection, Key Laboratory of Monitoring and Management of Crop Diseases and Pest Insects (Ministry of Agriculture), Nanjing Agricultural University, Nanjing 210095, China c Research and Development Centre of Biorational Pesticides, Northwest Agriculture and Forestry University, Yangling, China d CSIRO Land & Water Flagship, ACT, Australia e School of Biological Sciences, Australian National University, ACT, Australia f Cotton Catchment Communities CRC, Narrabri, NSW, Australia ARTICLE INFO ABSTRACT Article history: The low aqueous solubility and chiral complexity of synthetic pyrethroids, together with large differ- Received 31 October 2014 ences between isomers in their insecticidal potency, have hindered the development of meaningful assays Accepted 10 December 2014 of their metabolism and metabolic resistance to them. To overcome these problems, Shan and Hammock Available online 16 December 2014 (2001) [7] therefore developed fluorogenic and more water-soluble analogues of all the individual isomers of the commonly used Type 2 pyrethroids, cypermethrin and fenvalerate. -

Cholesterol Esterase from Porcine Pancreas
Cholesterol Esterase from porcine pancreas Catalog Number C9464 Storage Temperature –20 °C CAS RN 9026-00-0 Substrates: EC 3.1.1.13 cholesteryl esters17 triacylglycerol2 Synonyms: Bile salt activated lipase, sterol esterase, 4-nitrophenyl butyrate7 triolein9 carboxyl ester lipase, steryl-ester acylhydrolase 2-(diethylamino)-4H-3,1-benzoxazin-4-one18 2-(diethylamino)-4H-thieno[2,3-d]oxazin-4-one18 Product Description 7 Excess cholesterol is stored intracellularly as KM: 4-nitrophenyl butyrate cholesterol esters. Cholesterol esterase (CE) is a 0.37 mM (with taurocholate) reversible enzyme that can hydrolyze or synthesize 0.73 mM (without taurocholate) fatty acid esters of cholesterol and other sterols. Hydrolysis of water insoluble long chain fatty acid Activators: esters requires bile salt activation. Hydrolysis of water ethanol9 methanol9 soluble esters of short chain fatty acids and n-butanol9 sodium taurocholate10 lysophospholipids does not require activation by bile sodium cholate11 salts.1 Cholesterol esterase catalyzes the following reaction: Inhibitors: bisphenol A methacrylate12 Hg2+,15 Cholesterol Cholesterol Cholesterol diisopropylfluorophosphate13 enolase14 esters esterase + Fatty acid sodium fluoride15 phosphatidic acid16 phosphatidylcholine16 phosphatidylserine16 While found primarily in the pancreas and pancreatic 4-nitrophenyl-N-substituted carbamates11 fluid, it occurs in other tissues as well. In the bovine adrenal cortex, this reaction is one of the rate limiting Ki: steps in steroidogenesis, involving the release of 4-nitrophenyl-N-allyl -

Hydrolysis of Individual Isomers of Fluorogenic Pyrethroid Analogs by Mutant Carboxylesterases from Lucilia Cuprina
ARTICLE IN PRESS Insect Biochemistry and Molecular Biology Insect Biochemistry and Molecular Biology 37 (2007) 891–902 www.elsevier.com/locate/ibmb Hydrolysis of individual isomers of fluorogenic pyrethroid analogs by mutant carboxylesterases from Lucilia cuprina A.L. Devonshirea, R. Heidaria, H.Z. Huangb, B.D. Hammockb, R.J. Russella, J.G. Oakeshotta,Ã aCSIRO Entomology, GPO Box 1700, Canberra, ACT, 2601, Australia bDepartment of Entomology and Cancer Research Center, University of California, Davis, CA 95616, USA Received 28 November 2006; received in revised form 16 April 2007; accepted 17 April 2007 Abstract We previously showed that wild-type E3 carboxylesterase of Lucilia cuprina has high activity against Type 1 pyrethroids but much less for the bulkier, a-cyano containing Type 2 pyrethroids. Both Types have at least two optical centres and, at least for the Type 1 compounds, we found that wild-type E3 strongly prefers the less insecticidal configurations of the acyl group. However, substitutions to smaller residues at two sites in the acyl pocket of the enzyme substantially increased overall activity, particularly for the more insecticidal isomers. Here we extend these analyses to Type 2 pyrethroids by using fluorogenic analogs of all the diastereomers of cypermethrin and fenvalerate. Wild-type E3 hydrolysed some of these appreciably, but, again, not those corresponding to the most insecticidal isomers. Mutations in the leaving group pocket or oxyanion hole were again generally neutral or deleterious. However, the two sets of mutants in the acyl pocket again improved activity for the more insecticidal acyl group arrangements as well as for the more insecticidal configuration of the cyano moiety on the leaving group. -

Computational Modeling of Solvent Effects on Protein-Ligand
Commun. Comput. Phys. Vol. 13, No. 1, pp. 31-60 doi: 10.4208/cicp.130911.121011s January 2013 Computational Modeling of Solvent Effects on Protein-Ligand Interactions Using Fully Polarizable Continuum Model and Rational Drug Design Fang Zheng and Chang-Guo Zhan∗ Department of Pharmaceutical Sciences, College of Pharmacy, University of Kentucky, 789 South Limestone Street, Lexington, Kentucky 40536, USA. Received 13 September 2011; Accepted (in revised version) 12 October 2011 Available online 12 June 2012 Abstract. This is a brief review of the computational modeling of protein-ligand in- teractions using a recently developed fully polarizable continuum model (FPCM) and rational drug design. Computational modeling has become a powerful tool in under- standing detailed protein-ligand interactions at molecular level and in rational drug design. To study the binding of a protein with multiple molecular species of a ligand, one must accurately determine both the relative free energies of all of the molecular species in solution and the corresponding microscopic binding free energies for all of the molecular species binding with the protein. In this paper, we aim to provide a brief overview of the recent development in computational modeling of the solvent ef- fects on the detailed protein-ligand interactions involving multiple molecular species of a ligand related to rational drug design. In particular, we first briefly discuss the main challenges in computational modeling of the detailed protein-ligand interactions involving the multiple molecular species and then focus on the FPCM model and its applications. The FPCM method allows accurate determination of the solvent effects in the first-principles quantum mechanism (QM) calculations on molecules in solu- tion. -
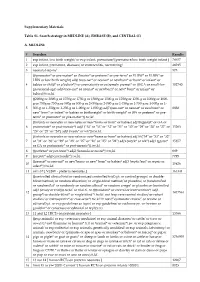
EMBASE (B), and CENTRAL (C) A. MEDLINE # Searches
Supplementary Materials Table S1. Search strategy in MEDLINE (A), EMBASE (B), and CENTRAL (C) A. MEDLINE # Searches Results 1 exp infant, low birth weight/ or exp infant, premature/ [premature/low birth weight infant ] 78657 2 exp infant, premature, diseases/ or enterocolitis, necrotizing/ 46015 3 neonatal sepsis/ 575 (((prematur* or pre-matur* or i?matur* or preterm* or pre-term* or VLBW* or ELBW* or LBW or low birth weight) adj6 (neo-nat* or neonat* or newborn* or born* or infant* or 4 babies or child* or p?ediatr*)) or prematurity or extremely premat* or ((SGA or small-for- 102740 gestational-age) adj6 (neo-nat* or neonat* or newborn* or new* born* or infant* or babies))).tw,kf. ((2000g or 2000-g or 1750g or 1750-g or 1500g or 1500-g or 1250g or 1250-g or 1000g or 1000- g or 750g or 750-g or 500g or 500-g or 2-000g or 2-000-g or 1-750g or 1-750-g or 1-500g or 1- 5 500-g or 1-250g or 1-250-g or 1-000g or 1-000-g) adj7 (neo-nat* or neonat* or newborn* or 8838 new* born* or infant* or babies or birthweight* or birth weight* or BW or preterm* or pre- term* or prematur* or pre-matur*)).tw,kf. ((infants or neonates or neo-nates or new*borns or born* or babies) adj18 (gestat* or GA or 6 postmenstr* or post-menstr*) adj3 ("34" or "33" or "32" or "31" or "30" or "29" or "28" or "27" or 15263 "26" or "25" or "24") adj3 (week* or wk*)).tw,kf. -

September, 2008
NIDA - Director's Report - September, 2008 NIDA Home > Publications > Director's Reports Director's Report to the National Advisory Council on Drug Abuse - September, 2008 Index Research Findings Cross-Divisional Research Basic Neurosciences Research Basic Behavioral Research Behavioral and Brain Development Research Clinical Neuroscience Research Epidemiology and Etiology Research Prevention Research Research on Behavioral and Combined Treatments for Drug Abuse Research on Pharmacotherapies for Drug Abuse Research on Medical Consequences of Drug Abuse and Co-Occurring Infections (HIV/AIDS, HCV) Services Research Clinical Trials Network Research International Research Intramural Research Program Activities Extramural Policy and Review Activities Congressional Affairs International Activities Meetings and Conferences Media and Education Activities Planned Meetings Publications Staff Highlights Grantee Honors https://archives.drugabuse.gov/DirReports/DirRep908/Default.html[11/17/16, 11:11:47 PM] NIDA - Director's Report - September, 2008 Archive Home | Accessibility | Privacy | FOIA (NIH) | Current NIDA Home Page The National Institute on Drug Abuse (NIDA) is part of the National Institutes of Health (NIH) , a component of the U.S. Department of Health and Human Services. Questions? _ See our Contact Information. https://archives.drugabuse.gov/DirReports/DirRep908/Default.html[11/17/16, 11:11:47 PM] NIDA - Director's Report - September, 2008 NIDA Home > Publications > Director's Reports > September, 2008 Index Director's Report to the National -

Cholesterol Esterase from Pseudomonas Sp
Cholesterol Esterase from Pseudomonas sp. Catalog Number C9281 Storage Temperature –20 C CAS RN 9026-00-0 Specific activity: 10,000 units/g protein EC 3.1.1.13 Synonyms: Bile salt activated lipase, sterol esterase, Unit definition: one unit will hydrolyze 1.0 mole of carboxyl ester lipase, steryl-ester acylhydrolase cholesteryl oleate to cholesterol and oleic acid per minute at pH 7.0 at 37 C in the presence of Product Description taurocholate. Excess cholesterol is stored intracellularly as cholesterol esters. Cholesterol esterase (CE) is a Cholesterol esterase is assayed spectrophotometrically reversible enzyme, with a molecular mass of in a 3.0 mL reaction mixture containing 287 mM 1 129 kDa, that can hydrolyze or synthesize fatty acid potassium phosphate, pH 7.0, 0.25% (w/v) taurocholic esters of cholesterol and other sterols. Hydrolysis of acid, 0.25% (w/v) cholic acid, 4-6 units peroxidase, water-insoluble long chain fatty acid esters requires bile 1.4 mM cholesteryl oleate, 1.7% (v/v) polyoxyethylene salt activation. Hydrolysis of water-soluble esters of 9-lauryl ether, 0.14% (w/v) NaCl, 0.083% (w/v) phenol, short chain fatty acids and lysophospholipids does not 0.03% (w/v) 4-aminoantipyrine, 1-1.5 units cholesterol 2 require activation by bile salts. Cholesterol esterase oxidase, and 0.013-0.143 unit cholesterol esterase. catalyzes the following reaction: Precautions and Disclaimer Cholesterol Cholesterol Cholesterol This product is for R&D use only, not for drug, esters esterase + Fatty acid household, or other uses. Please consult the Safety Data Sheet for information regarding hazards and safe In the bovine adrenal cortex, this reaction is one of the handling practices. -

Supplementary Material
Supplementary material Figure S1. Cluster analysis of the proteome profile based on qualitative data in low and high sugar conditions. Figure S2. Expression pattern of proteins under high and low sugar cultivation of Granulicella sp. WH15 a) All proteins identified in at least two out of three replicates (excluding on/off proteins). b) Only proteins with significant change t-test p=0.01. 2fold change is indicated by a red line. Figure S3. TigrFam roles of the differentially expressed proteins, excluding proteins with unknown function. Figure S4. General overview of up (red) and downregulated (blue) metabolic pathways based on KEGG analysis of proteome. Table S1. growth of strain Granulicella sp. WH15 in culture media supplemented with different carbon sources. Carbon Source Growth Pectin - Glycogen - Glucosamine - Cellulose - D-glucose + D-galactose + D-mannose + D-xylose + L-arabinose + L-rhamnose + D-galacturonic acid - Cellobiose + D-lactose + Sucrose + +=positive growth; -=No growth. Table S2. Total number of transcripts reads per sample in low and high sugar conditions. Sample ID Total Number of Reads Low sugar (1) 15,731,147 Low sugar (2) 12,624,878 Low sugar (3) 11,080,985 High sugar (1) 11,138,128 High sugar (2) 9,322,795 High sugar (3) 10,071,593 Table S3. Differentially up and down regulated transcripts in high sugar treatment. ORF Annotation Log2FC GWH15_14040 hypothetical protein 3.71 GWH15_06005 hypothetical protein 3.12 GWH15_00285 tRNA-Asn(gtt) 2.74 GWH15_06010 hypothetical protein 2.70 GWH15_14055 hypothetical protein 2.66 -

Identification and Characterization of a Novel Thermostable Pyrethroid
Fan et al. Microbial Cell Factories 2012, 11:33 http://www.microbialcellfactories.com/content/11/1/33 RESEARCH Open Access Identification and characterization of a novel thermostable pyrethroid-hydrolyzing enzyme isolated through metagenomic approach Xinjiong Fan1, Xiaolong Liu1,2, Rui Huang1 and Yuhuan Liu1* Abstract Background: Pyrethroid pesticides are broad-spectrum pest control agents in agricultural production. Both agricultural and residential usage is continuing to grow, leading to the development of insecticide resistance in the pest and toxic effects on a number of nontarget organisms. Thus, it is necessary to hunt suitable enzymes including hydrolases for degrading pesticide residues, which is an efficient “green” solution to biodegrade polluting chemicals. Although many pyrethroid esterases have consistently been purified and characterized from various resources including metagenomes and organisms, the thermostable pyrethroid esterases have not been reported up to the present. Results: In this study, we identified a novel pyrethroid-hydrolyzing enzyme Sys410 belonging to familyV esterases/ lipases with activity-based functional screening from Turban Basin metagenomic library. Sys410 contained 280 amino acids with a predicted molecular mass (Mr) of 30.8 kDa and was overexpressed in Escherichia coli BL21 (DE3) in soluble form. The optimum pH and temperature of the recombinant Sys410 were 6.5 and 55°C, respectively. The enzyme was stable in the pH range of 4.5-8.5 and at temperatures below 50°C. The activity of Sys410 decreased a little when stored at 4°C for 10 weeks, and the residual activity reached 94.1%. Even after incubation at 25°C for 10 weeks, it kept 68.3% of its activity.