The GAMESS-UK Electronic Structure Package: Algorithms, Developments and Applications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MODELLER 10.1 Manual
MODELLER A Program for Protein Structure Modeling Release 10.1, r12156 Andrej Saliˇ with help from Ben Webb, M.S. Madhusudhan, Min-Yi Shen, Guangqiang Dong, Marc A. Martı-Renom, Narayanan Eswar, Frank Alber, Maya Topf, Baldomero Oliva, Andr´as Fiser, Roberto S´anchez, Bozidar Yerkovich, Azat Badretdinov, Francisco Melo, John P. Overington, and Eric Feyfant email: modeller-care AT salilab.org URL https://salilab.org/modeller/ 2021/03/12 ii Contents Copyright notice xxi Acknowledgments xxv 1 Introduction 1 1.1 What is Modeller?............................................. 1 1.2 Modeller bibliography....................................... .... 2 1.3 Obtainingandinstallingtheprogram. .................... 3 1.4 Bugreports...................................... ............ 4 1.5 Method for comparative protein structure modeling by Modeller ................... 5 1.6 Using Modeller forcomparativemodeling. ... 8 1.6.1 Preparinginputfiles . ............. 8 1.6.2 Running Modeller ......................................... 9 2 Automated comparative modeling with AutoModel 11 2.1 Simpleusage ..................................... ............ 11 2.2 Moreadvancedusage............................... .............. 12 2.2.1 Including water molecules, HETATM residues, and hydrogenatoms .............. 12 2.2.2 Changing the default optimization and refinement protocol ................... 14 2.2.3 Getting a very fast and approximate model . ................. 14 2.2.4 Building a model from multiple templates . .................. 15 2.2.5 Buildinganallhydrogenmodel -

Integrated Tools for Computational Chemical Dynamics
UNIVERSITY OF MINNESOTA Integrated Tools for Computational Chemical Dynamics • Developpp powerful simulation methods and incorporate them into a user- friendly high-throughput integrated software su ite for c hem ica l d ynami cs New Models (Methods) → Modules → Integrated Tools UNIVERSITY OF MINNESOTA Development of new methods for the calculation of potential energy surface UNIVERSITY OF MINNESOTA Development of new simulation methods UNIVERSITY OF MINNESOTA Applications and Validations UNIVERSITY OF MINNESOTA Electronic Software structu re Dynamics ANT QMMM GCMC MN-GFM POLYRATE GAMESSPLUS MN-NWCHEMFM MN-QCHEMFM HONDOPLUS MN-GSM Interfaces GaussRate JaguarRate NWChemRate UNIVERSITY OF MINNESOTA Electronic Structure Software QMMM 1.3: QMMM is a computer program for combining quantum mechanics (QM) and molecular mechanics (MM). MN-GFM 3.0: MN-GFM is a module incorporating Minnesota DFT functionals into GAUSSIAN 03. MN-GSM 626.2:MN: MN-GSM is a module incorporating the SMx solvation models and other enhancements into GAUSSIAN 03. MN-NWCHEMFM 2.0:MN-NWCHEMFM is a module incorporating Minnesota DFT functionals into NWChem 505.0. MN-QCHEMFM 1.0: MN-QCHEMFM is a module incorporating Minnesota DFT functionals into Q-CHEM. GAMESSPLUS 4.8: GAMESSPLUS is a module incorporating the SMx solvation models and other enhancements into GAMESS. HONDOPLUS 5.1: HONDOPLUS is a computer program incorporating the SMx solvation models and other photochemical diabatic states into HONDO. UNIVERSITY OF MINNESOTA DiSftDynamics Software ANT 07: ANT is a molecular dynamics program for performing classical and semiclassical trajjyectory simulations for adiabatic and nonadiabatic processes. GCMC: GCMC is a Grand Canonical Monte Carlo (GCMC) module for the simulation of adsorption isotherms in heterogeneous catalysts. -

Supporting Information
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2020 Supporting Information How to Select Ionic Liquids as Extracting Agent Systematically? Special Case Study for Extractive Denitrification Process Shurong Gaoa,b,c,*, Jiaxin Jina,b, Masroor Abroc, Ruozhen Songc, Miao Hed, Xiaochun Chenc,* a State Key Laboratory of Alternate Electrical Power System with Renewable Energy Sources, North China Electric Power University, Beijing, 102206, China b Research Center of Engineering Thermophysics, North China Electric Power University, Beijing, 102206, China c Beijing Key Laboratory of Membrane Science and Technology & College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, PR China d Office of Laboratory Safety Administration, Beijing University of Technology, Beijing 100124, China * Corresponding author, Tel./Fax: +86-10-6443-3570, E-mail: [email protected], [email protected] 1 COSMO-RS Computation COSMOtherm allows for simple and efficient processing of large numbers of compounds, i.e., a database of molecular COSMO files; e.g. the COSMObase database. COSMObase is a database of molecular COSMO files available from COSMOlogic GmbH & Co KG. Currently COSMObase consists of over 2000 compounds including a large number of industrial solvents plus a wide variety of common organic compounds. All compounds in COSMObase are indexed by their Chemical Abstracts / Registry Number (CAS/RN), by a trivial name and additionally by their sum formula and molecular weight, allowing a simple identification of the compounds. We obtained the anions and cations of different ILs and the molecular structure of typical N-compounds directly from the COSMObase database in this manuscript. -

GROMACS: Fast, Flexible, and Free
GROMACS: Fast, Flexible, and Free DAVID VAN DER SPOEL,1 ERIK LINDAHL,2 BERK HESS,3 GERRIT GROENHOF,4 ALAN E. MARK,4 HERMAN J. C. BERENDSEN4 1Department of Cell and Molecular Biology, Uppsala University, Husargatan 3, Box 596, S-75124 Uppsala, Sweden 2Stockholm Bioinformatics Center, SCFAB, Stockholm University, SE-10691 Stockholm, Sweden 3Max-Planck Institut fu¨r Polymerforschung, Ackermannweg 10, D-55128 Mainz, Germany 4Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 4, NL-9747 AG Groningen, The Netherlands Received 12 February 2005; Accepted 18 March 2005 DOI 10.1002/jcc.20291 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: This article describes the software suite GROMACS (Groningen MAchine for Chemical Simulation) that was developed at the University of Groningen, The Netherlands, in the early 1990s. The software, written in ANSI C, originates from a parallel hardware project, and is well suited for parallelization on processor clusters. By careful optimization of neighbor searching and of inner loop performance, GROMACS is a very fast program for molecular dynamics simulation. It does not have a force field of its own, but is compatible with GROMOS, OPLS, AMBER, and ENCAD force fields. In addition, it can handle polarizable shell models and flexible constraints. The program is versatile, as force routines can be added by the user, tabulated functions can be specified, and analyses can be easily customized. Nonequilibrium dynamics and free energy determinations are incorporated. Interfaces with popular quantum-chemical packages (MOPAC, GAMES-UK, GAUSSIAN) are provided to perform mixed MM/QM simula- tions. The package includes about 100 utility and analysis programs. -

Chem Compute Quickstart
Chem Compute Quickstart Chem Compute is maintained by Mark Perri at Sonoma State University and hosted on Jetstream at Indiana University. The Chem Compute URL is https://chemcompute.org/. We will use Chem Compute as the frontend for running electronic structure calculations with The General Atomic and Molecular Electronic Structure System, GAMESS (http://www.msg.ameslab.gov/gamess/). Chem Compute also provides access to other computational chemistry resources including PSI4 and the molecular dynamics packages TINKER and NAMD, though we will not be using those resource at this time. Follow this link, https://chemcompute.org/gamess/submit, to directly access the Chem Compute GAMESS guided submission interface. If you are a returning Chem Computer user, please log in now. If your University is part of the InCommon Federation you can log in without registering by clicking Login then "Log In with Google or your University" – select your University from the dropdown list. Otherwise if this is your first time working with Chem Compute, please register as a Chem Compute user by clicking the “Register” link in the top-right corner of the page. This gives you access to all of the computational resources available at ChemCompute.org and will allow you to maintain copies of your calculations in your user “Dashboard” that you can refer to later. Registering also helps track usage and obtain the resources needed to continue providing its service. When logged in with the GAMESS-Submit tabs selected, an instruction section appears on the left side of the page with instructions for several different kinds of calculations. -

Popular GPU-Accelerated Applications
LIFE & MATERIALS SCIENCES GPU-ACCELERATED APPLICATIONS | CATALOG | AUG 12 LIFE & MATERIALS SCIENCES APPLICATIONS CATALOG Application Description Supported Features Expected Multi-GPU Release Status Speed Up* Support Bioinformatics BarraCUDA Sequence mapping software Alignment of short sequencing 6-10x Yes Available now reads Version 0.6.2 CUDASW++ Open source software for Smith-Waterman Parallel search of Smith- 10-50x Yes Available now protein database searches on GPUs Waterman database Version 2.0.8 CUSHAW Parallelized short read aligner Parallel, accurate long read 10x Yes Available now aligner - gapped alignments to Version 1.0.40 large genomes CATALOG GPU-BLAST Local search with fast k-tuple heuristic Protein alignment according to 3-4x Single Only Available now blastp, multi cpu threads Version 2.2.26 GPU-HMMER Parallelized local and global search with Parallel local and global search 60-100x Yes Available now profile Hidden Markov models of Hidden Markov Models Version 2.3.2 mCUDA-MEME Ultrafast scalable motif discovery algorithm Scalable motif discovery 4-10x Yes Available now based on MEME algorithm based on MEME Version 3.0.12 MUMmerGPU A high-throughput DNA sequence alignment Aligns multiple query sequences 3-10x Yes Available now LIFE & MATERIALS& LIFE SCIENCES APPLICATIONS program against reference sequence in Version 2 parallel SeqNFind A GPU Accelerated Sequence Analysis Toolset HW & SW for reference 400x Yes Available now assembly, blast, SW, HMM, de novo assembly UGENE Opensource Smith-Waterman for SSE/CUDA, Fast short -

Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development
processes Review Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development Outi M. H. Salo-Ahen 1,2,* , Ida Alanko 1,2, Rajendra Bhadane 1,2 , Alexandre M. J. J. Bonvin 3,* , Rodrigo Vargas Honorato 3, Shakhawath Hossain 4 , André H. Juffer 5 , Aleksei Kabedev 4, Maija Lahtela-Kakkonen 6, Anders Støttrup Larsen 7, Eveline Lescrinier 8 , Parthiban Marimuthu 1,2 , Muhammad Usman Mirza 8 , Ghulam Mustafa 9, Ariane Nunes-Alves 10,11,* , Tatu Pantsar 6,12, Atefeh Saadabadi 1,2 , Kalaimathy Singaravelu 13 and Michiel Vanmeert 8 1 Pharmaceutical Sciences Laboratory (Pharmacy), Åbo Akademi University, Tykistökatu 6 A, Biocity, FI-20520 Turku, Finland; ida.alanko@abo.fi (I.A.); rajendra.bhadane@abo.fi (R.B.); parthiban.marimuthu@abo.fi (P.M.); atefeh.saadabadi@abo.fi (A.S.) 2 Structural Bioinformatics Laboratory (Biochemistry), Åbo Akademi University, Tykistökatu 6 A, Biocity, FI-20520 Turku, Finland 3 Faculty of Science-Chemistry, Bijvoet Center for Biomolecular Research, Utrecht University, 3584 CH Utrecht, The Netherlands; [email protected] 4 Swedish Drug Delivery Forum (SDDF), Department of Pharmacy, Uppsala Biomedical Center, Uppsala University, 751 23 Uppsala, Sweden; [email protected] (S.H.); [email protected] (A.K.) 5 Biocenter Oulu & Faculty of Biochemistry and Molecular Medicine, University of Oulu, Aapistie 7 A, FI-90014 Oulu, Finland; andre.juffer@oulu.fi 6 School of Pharmacy, University of Eastern Finland, FI-70210 Kuopio, Finland; maija.lahtela-kakkonen@uef.fi (M.L.-K.); tatu.pantsar@uef.fi -

An Efficient Hardware-Software Approach to Network Fault Tolerance with Infiniband
An Efficient Hardware-Software Approach to Network Fault Tolerance with InfiniBand Abhinav Vishnu1 Manoj Krishnan1 and Dhableswar K. Panda2 Pacific Northwest National Lab1 The Ohio State University2 Outline Introduction Background and Motivation InfiniBand Network Fault Tolerance Primitives Hybrid-IBNFT Design Hardware-IBNFT and Software-IBNFT Performance Evaluation of Hybrid-IBNFT Micro-benchmarks and NWChem Conclusions and Future Work Introduction Clusters are observing a tremendous increase in popularity Excellent price to performance ratio 82% supercomputers are clusters in June 2009 TOP500 rankings Multiple commodity Interconnects have emerged during this trend InfiniBand, Myrinet, 10GigE …. InfiniBand has become popular Open standard and high performance Various topologies have emerged for interconnecting InfiniBand Fat tree is the predominant topology TACC Ranger, PNNL Chinook 3 Typical InfiniBand Fat Tree Configurations 144-Port Switch Block Diagram Multiple leaf and spine blocks 12 Spine Available in 144, 288 and 3456 port Blocks combinations 12 Leaf Blocks Multiple paths are available between nodes present on different switch blocks Oversubscribed configurations are becoming popular 144-Port Switch Block Diagram Better cost to performance ratio 12 Spine Blocks 12 Spine 12 Spine Blocks Blocks 12 Leaf 12 Leaf Blocks Blocks 4 144-Port Switch Block Diagram 144-Port Switch Block Diagram Network Faults Links/Switches/Adapters may fail with reduced MTBF (Mean time between failures) Fortunately, InfiniBand provides mechanisms to handle -

Computer-Assisted Catalyst Development Via Automated Modelling of Conformationally Complex Molecules
www.nature.com/scientificreports OPEN Computer‑assisted catalyst development via automated modelling of conformationally complex molecules: application to diphosphinoamine ligands Sibo Lin1*, Jenna C. Fromer2, Yagnaseni Ghosh1, Brian Hanna1, Mohamed Elanany3 & Wei Xu4 Simulation of conformationally complicated molecules requires multiple levels of theory to obtain accurate thermodynamics, requiring signifcant researcher time to implement. We automate this workfow using all open‑source code (XTBDFT) and apply it toward a practical challenge: diphosphinoamine (PNP) ligands used for ethylene tetramerization catalysis may isomerize (with deleterious efects) to iminobisphosphines (PPNs), and a computational method to evaluate PNP ligand candidates would save signifcant experimental efort. We use XTBDFT to calculate the thermodynamic stability of a wide range of conformationally complex PNP ligands against isomeriation to PPN (ΔGPPN), and establish a strong correlation between ΔGPPN and catalyst performance. Finally, we apply our method to screen novel PNP candidates, saving signifcant time by ruling out candidates with non‑trivial synthetic routes and poor expected catalytic performance. Quantum mechanical methods with high energy accuracy, such as density functional theory (DFT), can opti- mize molecular input structures to a nearby local minimum, but calculating accurate reaction thermodynamics requires fnding global minimum energy structures1,2. For simple molecules, expert intuition can identify a few minima to focus study on, but an alternative approach must be considered for more complex molecules or to eventually fulfl the dream of autonomous catalyst design 3,4: the potential energy surface must be frst surveyed with a computationally efcient method; then minima from this survey must be refned using slower, more accurate methods; fnally, for molecules possessing low-frequency vibrational modes, those modes need to be treated appropriately to obtain accurate thermodynamic energies 5–7. -

Starting SCF Calculations by Superposition of Atomic Densities
Starting SCF Calculations by Superposition of Atomic Densities J. H. VAN LENTHE,1 R. ZWAANS,1 H. J. J. VAN DAM,2 M. F. GUEST2 1Theoretical Chemistry Group (Associated with the Department of Organic Chemistry and Catalysis), Debye Institute, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands 2CCLRC Daresbury Laboratory, Daresbury WA4 4AD, United Kingdom Received 5 July 2005; Accepted 20 December 2005 DOI 10.1002/jcc.20393 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: We describe the procedure to start an SCF calculation of the general type from a sum of atomic electron densities, as implemented in GAMESS-UK. Although the procedure is well known for closed-shell calculations and was already suggested when the Direct SCF procedure was proposed, the general procedure is less obvious. For instance, there is no need to converge the corresponding closed-shell Hartree–Fock calculation when dealing with an open-shell species. We describe the various choices and illustrate them with test calculations, showing that the procedure is easier, and on average better, than starting from a converged minimal basis calculation and much better than using a bare nucleus Hamiltonian. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 926–932, 2006 Key words: SCF calculations; atomic densities Introduction hrstuhl fur Theoretische Chemie, University of Kahrlsruhe, Tur- bomole; http://www.chem-bio.uni-karlsruhe.de/TheoChem/turbo- Any quantum chemical calculation requires properly defined one- mole/),12 GAMESS(US) (Gordon Research Group, GAMESS, electron orbitals. These orbitals are in general determined through http://www.msg.ameslab.gov/GAMESS/GAMESS.html, 2005),13 an iterative Hartree–Fock (HF) or Density Functional (DFT) pro- Spartan (Wavefunction Inc., SPARTAN: http://www.wavefun. -

Parameterizing a Novel Residue
University of Illinois at Urbana-Champaign Luthey-Schulten Group, Department of Chemistry Theoretical and Computational Biophysics Group Computational Biophysics Workshop Parameterizing a Novel Residue Rommie Amaro Brijeet Dhaliwal Zaida Luthey-Schulten Current Editors: Christopher Mayne Po-Chao Wen February 2012 CONTENTS 2 Contents 1 Biological Background and Chemical Mechanism 4 2 HisH System Setup 7 3 Testing out your new residue 9 4 The CHARMM Force Field 12 5 Developing Topology and Parameter Files 13 5.1 An Introduction to a CHARMM Topology File . 13 5.2 An Introduction to a CHARMM Parameter File . 16 5.3 Assigning Initial Values for Unknown Parameters . 18 5.4 A Closer Look at Dihedral Parameters . 18 6 Parameter generation using SPARTAN (Optional) 20 7 Minimization with new parameters 32 CONTENTS 3 Introduction Molecular dynamics (MD) simulations are a powerful scientific tool used to study a wide variety of systems in atomic detail. From a standard protein simulation, to the use of steered molecular dynamics (SMD), to modelling DNA-protein interactions, there are many useful applications. With the advent of massively parallel simulation programs such as NAMD2, the limits of computational anal- ysis are being pushed even further. Inevitably there comes a time in any molecular modelling scientist’s career when the need to simulate an entirely new molecule or ligand arises. The tech- nique of determining new force field parameters to describe these novel system components therefore becomes an invaluable skill. Determining the correct sys- tem parameters to use in conjunction with the chosen force field is only one important aspect of the process. -
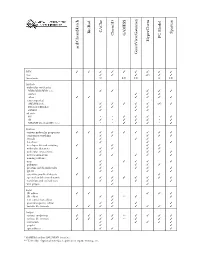
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only.