7Th Conference on Beneficial Microbes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phylogenetic Rooting Using Minimal Ancestor Deviation
Phylogenetic rooting using minimal ancestor deviation Fernando D. K. Tria1, Giddy Landan1*, Tal Dagan Genomic Microbiology Group, Institute of General Microbiology, Kiel University, Kiel, Germany 1 Equally contributed. * Corresponding author: [email protected] This preprint PDF is the revised manuscript as submitted to Nature Ecology & Evolution on 30-Mar-2017. It includes both the main text and the supplementary information. The final version published in Nature Ecology & Evolution on 19-Jun-2017 (and submitted on 08-May-2017), is here: https://www.nature.com/articles/s41559-017-0193 Readers without subscription to NatE&E can see a read-only (no save or print) version here: http://rdcu.be/tywU The main difference between the versions is that the ‘Detailed Algorithm’ section is part of the main text 'Methods' section in the NatE&E final version, but is part of the supplementary information of this preprint version. Be sure to page beyond the references to see it. 1 Abstract Ancestor-descendent relations play a cardinal role in evolutionary theory. Those relatio ns are determined by rooting phylogenetic trees. Existing rooting methods are hampered by evolutionary rate heterogeneity or the unavailability of auxiliary phylogenetic information. We present a novel rooting approach, the minimal ancestor deviation (MAD) method, which embraces heterotachy by utilizing all pairwise topological and metric information in unrooted trees. We demonstrate the method in comparison to existing rooting methods by the analysis of phylogenies from eukaryotes and prokaryotes. MAD correctly recovers the kno wn root of eukaryotes and uncovers evidence for cyanobacteria origins in the ocean. MAD is more robust and co nsistent than existing methods, provides measures of the root inference quality, and is applicable to any tree with branch lengths. -

Contrasting Environmental Preferences of Photosynthetic and Non-Photosynthetic Soil Cyanobacteria Across the Globe
Received: 31 October 2019 | Revised: 8 July 2020 | Accepted: 20 July 2020 DOI: 10.1111/geb.13173 RESEARCH PAPER Contrasting environmental preferences of photosynthetic and non-photosynthetic soil cyanobacteria across the globe Concha Cano-Díaz1 | Fernando T. Maestre2,3 | David J. Eldridge4 | Brajesh K. Singh5,6 | Richard D. Bardgett7 | Noah Fierer8,9 | Manuel Delgado-Baquerizo10 1Departamento de Biología, Geología, Física y Química Inorgánica, Escuela Superior de Ciencias Experimentales y Tecnología, Universidad Rey Juan Carlos, Móstoles, 28933, Spain 2Instituto Multidisciplinar para el Estudio del Medio “Ramón Margalef”, Universidad de Alicante, Edificio Nuevos Institutos, Carretera de San Vicente del Raspeig s/n, San Vicente del Raspeig, 03690, Spain 3Departamento de Ecología, Universidad de Alicante, Carretera de San Vicente del Raspeig s/n, San Vicente del Raspeig, 03690, Spain 4Centre for Ecosystem Science, School of Biological, Earth and Environmental Sciences, University of New South Wales, Sydney, New South Wales, Australia 5Global Centre for Land-Based Innovation, University of Western Sydney, Penrith, New South Wales, Australia 6Hawkesbury Institute for the Environment, University of Western Sydney, Penrith, New South Wales, Australia 7Department of Earth and Environmental Sciences, The University of Manchester, Manchester, UK 8Department of Ecology and Evolutionary Biology, University of Colorado, Boulder, CO, USA 9Cooperative Institute for Research in Environmental Sciences, University of Colorado, Boulder, Colorado, USA 10Departamento de Sistemas Físicos, Químicos y Naturales, Universidad Pablo de Olavide, Sevilla, 41013, Spain Correspondence Concha Cano-Díaz, Departamento de Abstract Biología, Geología, Física y Química Aim: Cyanobacteria have shaped the history of life on Earth and continue to play Inorgánica, Escuela Superior de Ciencias Experimentales y Tecnología, Universidad important roles as carbon and nitrogen fixers in terrestrial ecosystems. -

A Conserved Role for a GATA Transcription Factor in Regulating Epithelial Innate Immune Responses
A conserved role for a GATA transcription factor in regulating epithelial innate immune responses Michael Shapira*, Brigham J. Hamlin*, Jiming Rong*, Karen Chen*, Michal Ronen*, and Man-Wah Tan*†‡ Departments of *Genetics and †Microbiology and Immunology, Stanford University School of Medicine, Stanford, CA 94305 Edited by Frederick M. Ausubel, Harvard Medical School, Boston, MA, and approved July 10, 2006 (received for review April 26, 2006) Innate immunity is an ancient and conserved defense mechanism. adulthood (17). Although GATA transcription factors are ex- Although host responses toward various pathogens have been pressed during adulthood or in mature tissues, in other animals as delineated, how these responses are orchestrated in a whole well, very little is known about their postdevelopmental roles. animal is less understood. Through an unbiased genome-wide Here we show that in both C. elegans and human cells, an study performed in Caenorhabditis elegans, we identified a con- endodermal GATA transcription factor has a postdevelopmental served function for endodermal GATA transcription factors in role in local defense against the human Gram-negative bacterial regulating local epithelial innate immune responses. Gene expres- pathogen Pseudomonas aeruginosa. Using whole-genome gene ex- sion and functional RNAi-based analyses identified the tissue- pression and functional analyses in C. elegans, we identified the specific GATA transcription factor ELT-2 as a major regulator of an GATA transcription factor ELT-2 as a major regulator of an early early intestinal protective response to infection with the human protective response to P. aeruginosa. We found that the human bacterial pathogen Pseudomonas aeruginosa. In the adult worm, homolog, GATA6, protects lung epithelial cells from a similar ELT-2 is required specifically for infection responses and survival on infection, describing a conserved role for GATA transcription pathogen but makes no significant contribution to gene expression factors in mediating local epithelial responses to infection. -

The Mysterious Orphans of Mycoplasmataceae
The mysterious orphans of Mycoplasmataceae Tatiana V. Tatarinova1,2*, Inna Lysnyansky3, Yuri V. Nikolsky4,5,6, and Alexander Bolshoy7* 1 Children’s Hospital Los Angeles, Keck School of Medicine, University of Southern California, Los Angeles, 90027, California, USA 2 Spatial Science Institute, University of Southern California, Los Angeles, 90089, California, USA 3 Mycoplasma Unit, Division of Avian and Aquatic Diseases, Kimron Veterinary Institute, POB 12, Beit Dagan, 50250, Israel 4 School of Systems Biology, George Mason University, 10900 University Blvd, MSN 5B3, Manassas, VA 20110, USA 5 Biomedical Cluster, Skolkovo Foundation, 4 Lugovaya str., Skolkovo Innovation Centre, Mozhajskij region, Moscow, 143026, Russian Federation 6 Vavilov Institute of General Genetics, Moscow, Russian Federation 7 Department of Evolutionary and Environmental Biology and Institute of Evolution, University of Haifa, Israel 1,2 [email protected] 3 [email protected] 4-6 [email protected] 7 [email protected] 1 Abstract Background: The length of a protein sequence is largely determined by its function, i.e. each functional group is associated with an optimal size. However, comparative genomics revealed that proteins’ length may be affected by additional factors. In 2002 it was shown that in bacterium Escherichia coli and the archaeon Archaeoglobus fulgidus, protein sequences with no homologs are, on average, shorter than those with homologs [1]. Most experts now agree that the length distributions are distinctly different between protein sequences with and without homologs in bacterial and archaeal genomes. In this study, we examine this postulate by a comprehensive analysis of all annotated prokaryotic genomes and focusing on certain exceptions. -

Table S4. Phylogenetic Distribution of Bacterial and Archaea Genomes in Groups A, B, C, D, and X
Table S4. Phylogenetic distribution of bacterial and archaea genomes in groups A, B, C, D, and X. Group A a: Total number of genomes in the taxon b: Number of group A genomes in the taxon c: Percentage of group A genomes in the taxon a b c cellular organisms 5007 2974 59.4 |__ Bacteria 4769 2935 61.5 | |__ Proteobacteria 1854 1570 84.7 | | |__ Gammaproteobacteria 711 631 88.7 | | | |__ Enterobacterales 112 97 86.6 | | | | |__ Enterobacteriaceae 41 32 78.0 | | | | | |__ unclassified Enterobacteriaceae 13 7 53.8 | | | | |__ Erwiniaceae 30 28 93.3 | | | | | |__ Erwinia 10 10 100.0 | | | | | |__ Buchnera 8 8 100.0 | | | | | | |__ Buchnera aphidicola 8 8 100.0 | | | | | |__ Pantoea 8 8 100.0 | | | | |__ Yersiniaceae 14 14 100.0 | | | | | |__ Serratia 8 8 100.0 | | | | |__ Morganellaceae 13 10 76.9 | | | | |__ Pectobacteriaceae 8 8 100.0 | | | |__ Alteromonadales 94 94 100.0 | | | | |__ Alteromonadaceae 34 34 100.0 | | | | | |__ Marinobacter 12 12 100.0 | | | | |__ Shewanellaceae 17 17 100.0 | | | | | |__ Shewanella 17 17 100.0 | | | | |__ Pseudoalteromonadaceae 16 16 100.0 | | | | | |__ Pseudoalteromonas 15 15 100.0 | | | | |__ Idiomarinaceae 9 9 100.0 | | | | | |__ Idiomarina 9 9 100.0 | | | | |__ Colwelliaceae 6 6 100.0 | | | |__ Pseudomonadales 81 81 100.0 | | | | |__ Moraxellaceae 41 41 100.0 | | | | | |__ Acinetobacter 25 25 100.0 | | | | | |__ Psychrobacter 8 8 100.0 | | | | | |__ Moraxella 6 6 100.0 | | | | |__ Pseudomonadaceae 40 40 100.0 | | | | | |__ Pseudomonas 38 38 100.0 | | | |__ Oceanospirillales 73 72 98.6 | | | | |__ Oceanospirillaceae -

Within-Arctic Horizontal Gene Transfer As a Driver of Convergent Evolution in Distantly Related 1 Microalgae 2 Richard G. Do
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.31.454568; this version posted August 2, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Within-Arctic horizontal gene transfer as a driver of convergent evolution in distantly related 2 microalgae 3 Richard G. Dorrell*+1,2, Alan Kuo3*, Zoltan Füssy4, Elisabeth Richardson5,6, Asaf Salamov3, Nikola 4 Zarevski,1,2,7 Nastasia J. Freyria8, Federico M. Ibarbalz1,2,9, Jerry Jenkins3,10, Juan Jose Pierella 5 Karlusich1,2, Andrei Stecca Steindorff3, Robyn E. Edgar8, Lori Handley10, Kathleen Lail3, Anna Lipzen3, 6 Vincent Lombard11, John McFarlane5, Charlotte Nef1,2, Anna M.G. Novák Vanclová1,2, Yi Peng3, Chris 7 Plott10, Marianne Potvin8, Fabio Rocha Jimenez Vieira1,2, Kerrie Barry3, Joel B. Dacks5, Colomban de 8 Vargas2,12, Bernard Henrissat11,13, Eric Pelletier2,14, Jeremy Schmutz3,10, Patrick Wincker2,14, Chris 9 Bowler1,2, Igor V. Grigoriev3,15, and Connie Lovejoy+8 10 11 1 Institut de Biologie de l'ENS (IBENS), Département de Biologie, École Normale Supérieure, CNRS, 12 INSERM, Université PSL, 75005 Paris, France 13 2CNRS Research Federation for the study of Global Ocean Systems Ecology and Evolution, 14 FR2022/Tara Oceans GOSEE, 3 rue Michel-Ange, 75016 Paris, France 15 3 US Department of Energy Joint Genome Institute, Lawrence Berkeley National Laboratory, 1 16 Cyclotron Road, Berkeley, -

Correlation Between the Oral Microbiome and Brain Resting State Connectivity in Smokers
bioRxiv preprint doi: https://doi.org/10.1101/444612; this version posted October 16, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Correlation between the oral microbiome and brain resting state connectivity in smokers Dongdong Lin1, Kent Hutchison2, Salvador Portillo3, Victor Vegara1, Jarod Ellingson2, Jingyu Liu1,3, Amanda Carroll-Portillo3,* ,Vince D. Calhoun1,3,* 1The Mind Research Network, Albuquerque, New Mexico, 87106 2University of Colorado Boulder, Boulder, CO 3University of New Mexico, Department of Electrical and Computer Engineering, Albuquerque, New Mexico, 87106 * authors contributed equally to the work. Abstract Recent studies have shown a critical role for the gastrointestinal microbiome in brain and behavior via a complex gut–microbiome–brain axis, however, the influence of the oral microbiome in neurological processes is much less studied, especially in response to the stimuli in the oral microenvironment such as smoking. Additionally, given the complex structural and functional networks in brain system, our knowledge about the relationship between microbiome and brain functions on specific brain circuits is still very limited. In this pilot work, we leverage next generation microbial sequencing with functional MRI techniques to enable the delineation of microbiome-brain network links as well as their relations to cigarette smoking. Thirty smokers and 30 age- and sex- matched non-smokers were recruited for measuring both microbial community and brain functional networks. Statistical analyses were performed to demonstrate the influence of smoking on: the taxonomy and abundance of the constituents within the oral microbial community, brain functional network connectivity, and associations between microbial shifts and the brain signaling network. -

Supplemental Materials Adaptations of Atribacteria to Life In
1 Supplemental Materials 2 Adaptations of Atribacteria to life in methane hydrates: hot traits for cold life 3 Authors: Jennifer B. Glass1*, Piyush Ranjan2*, Cecilia B. Kretz1#, Brook L. Nunn3, Abigail M. Johnson1, James McManus4, Frank J. Stewart2 4 Affiliations: 5 1School of Earth and Atmospheric Sciences, Georgia Institute of Technology, Atlanta, GA, USA 6 2School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, USA 7 3Department of Genome Sciences, University of Washington, Seattle, WA 8 4Bigelow Laboratory for Ocean Sciences, East Boothbay, ME, USA 9 10 #Now at: Division of Bacterial Diseases, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention, 11 Atlanta, Georgia, USA 12 *Correspondence to: [email protected] 13 Dedication: To Katrina Edwards 14 1 15 Table S1. Additional geochemical data for ODP Site 1244 at Hydrate Ridge drilled on IODP Leg 204. 16 See Fig. 1 for methane, sulfate, manganese, iron, and iodide concentrations. 17 Depth reactive reactive Hole (mbsf) %TC %TN %TS %TIC %TOC C:N %CaCO3 Fe (%) Mn (%) C1H2 1.95/2.25 2.07 0.2 0.42 0.17 1.9 11.05 1.46 0.38 0.002 C1H3 3.45/3.75 1.88 0.14 0.55 0.70 1.2 9.85 5.81 0.45 0.004 F2H4 8.6 1.54 0.17 0.39 0.44 1.1 7.55 3.66 0.57 0.005 F3H4 18.1 1.55 0.24 0.64 0.08 1.5 7.14 0.68 0.80 0.004 C3H4 20.69 1.22 0.18 0.22 0.08 1.1 7.40 0.65 1.10 0.004 E10H5 68.55 1.71 0.22 0.42 0.13 1.6 8.38 1.08 0.51 0.003 E19H5 138.89 1.42 0.20 0.07 0.51 0.9 5.32 4.23 1.41 0.012 18 2 19 Table S2. -
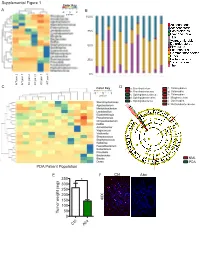
Mental Figure 1 Color Key a -2 0 2 B Z-Score 100%
Supplemental Figure 1 Color Key A -2 0 2 B z-score 100% 75% 50% 25% 0% KC pan 1 WT pan 3 WT KC pan 3 WT pan 2 WT pan 1 WT KC pan 2 C Color Key D a: Brevibacterium f: Chlamydiales b: Brevibacteriaceae g: Chlamydiia -3 0 3 z-score c: Sphingobacteriaceae h: Chlamydiae d: Sphingobacteriales i: Mogibacterium e: Sphingobacteriia j: Oscillospira k: Methylobacteriaceae NML Control Microb.-entrained MΦ PDA PDA Patient Population Control Microb.-entrained MΦ + Myd88i E F Ctrl Abx 350 * 300 250 200 150 40X 100 Tumor weight (mg) 50 0 x Ctrl Ab Supplemental Figure 2 A KC WT B ** * Actinobacteria * ** Bacteroidetes Cyanobacteria Deferribacteres * Firmicutes Proteobacteria % Relative abundance TM7 Others Time(wks) 3 9 13 16 20 24 28 32 36 3 9 13 16 20 24 28 32 36 Alpha Diversity Measure C E 60 KC WT 40 20 B. pseudolongum B. animalis 60 5 KC WT 0 B. adolescentis 40% Rel. abundance 3 9 13 16 20 24 28 32 36 3 9 13 16 20 24 28 32 36 20 Age (weeks) B. pseudolongum B. animalis 5 0 B. adolescentis % Rel. abundance 3 9 13 16 20 24 28 32 36 3 9 13 16 20 24 28 32 36 F Age (weeks) Week 3 Week 9 Week 13 p=0.678 p=0.02 p=0.385 Time(wks) 3 9 24 20 13 16 D 28 32 36 Week 13 KC WT Firmicutes; Ruminococcus Firmicutes; Dehalobacterium Alpha Diversity Measure Firmicutes; Oscillospira Bacteroidates; Odoribacter Axis.2 [12.7%] Actinobacteria; Bifidobacterium Axis.2 [23.8%] Axis.2 [24.7%] Week 16 Bacteroidetes; Bacteroidales Axis.1 [80.8%] Axis.1 [65.4%] Axis.1 [49.6%] Actinobacteria; Bifidobacterium Week 16 Week 20 Week 24 Week 20 p=0.339 p=0.036 p=0.021 Firmicutes; Dehalobacterium -

Exploring the Cockatiel (Nymphicus Hollandicus) Fecal Microbiome, Bacterial Inhabitants of a Worldwide Pet
Exploring the cockatiel (Nymphicus hollandicus) fecal microbiome, bacterial inhabitants of a worldwide pet Luis David Alcaraz1, Apolinar M. Hernández2 and Mariana Peimbert2 1 Laboratorio Nacional de Ciencias de la Sostenibilidad, Instituto de Ecología, Universidad Nacional Autonóma de México, Mexico City, Mexico 2 Departamento de Ciencias Naturales, Unidad Cuajimalpa, Universidad Autónoma Metropolitana, Mexico City, Mexico ABSTRACT Background. Cockatiels (Nymphicus hollandicus) were originally endemic to Australia; now they are popular pets with a global distribution. It is now possible to conduct detailed molecular studies on cultivable and uncultivable bacteria that are part of the intestinal microbiome of healthy animals. These studies show that bacteria are an essential part of the metabolic capacity of animals. There are few studies on bird microbiomes and, to the best of our knowledge, this is the first report on the cockatiel microbiome. Methods. In this paper, we analyzed the gut microbiome from fecal samples of three healthy adult cockatiels by massive sequencing of the 16S rRNA gene. Additionally, we compared the cockatiel fecal microbiomes with those of other bird species, including poultry and wild birds. Results. The vast majority of the bacteria found in cockatiels were Firmicutes, while Proteobacteria and Bacteroidetes were poorly represented. A total of 19,280 different OTUs were detected, of which 8,072 belonged to the Erysipelotrichaceae family. Discussion. It is relevant to study cockatiel the microbiomes of cockatiels owing to their wide geographic distribution and close human contact. This study serves as a reference for cockatiel bacterial diversity. Despite the large OTU numbers, the diversity is not even Submitted 14 July 2016 Accepted 28 November 2016 and is dominated by Firmicutes of the Erysipelotrichaceae family. -

Compile.Xlsx
Silva OTU GS1A % PS1B % Taxonomy_Silva_132 otu0001 0 0 2 0.05 Bacteria;Acidobacteria;Acidobacteria_un;Acidobacteria_un;Acidobacteria_un;Acidobacteria_un; otu0002 0 0 1 0.02 Bacteria;Acidobacteria;Acidobacteriia;Solibacterales;Solibacteraceae_(Subgroup_3);PAUC26f; otu0003 49 0.82 5 0.12 Bacteria;Acidobacteria;Aminicenantia;Aminicenantales;Aminicenantales_fa;Aminicenantales_ge; otu0004 1 0.02 7 0.17 Bacteria;Acidobacteria;AT-s3-28;AT-s3-28_or;AT-s3-28_fa;AT-s3-28_ge; otu0005 1 0.02 0 0 Bacteria;Acidobacteria;Blastocatellia_(Subgroup_4);Blastocatellales;Blastocatellaceae;Blastocatella; otu0006 0 0 2 0.05 Bacteria;Acidobacteria;Holophagae;Subgroup_7;Subgroup_7_fa;Subgroup_7_ge; otu0007 1 0.02 0 0 Bacteria;Acidobacteria;ODP1230B23.02;ODP1230B23.02_or;ODP1230B23.02_fa;ODP1230B23.02_ge; otu0008 1 0.02 15 0.36 Bacteria;Acidobacteria;Subgroup_17;Subgroup_17_or;Subgroup_17_fa;Subgroup_17_ge; otu0009 9 0.15 41 0.99 Bacteria;Acidobacteria;Subgroup_21;Subgroup_21_or;Subgroup_21_fa;Subgroup_21_ge; otu0010 5 0.08 50 1.21 Bacteria;Acidobacteria;Subgroup_22;Subgroup_22_or;Subgroup_22_fa;Subgroup_22_ge; otu0011 2 0.03 11 0.27 Bacteria;Acidobacteria;Subgroup_26;Subgroup_26_or;Subgroup_26_fa;Subgroup_26_ge; otu0012 0 0 1 0.02 Bacteria;Acidobacteria;Subgroup_5;Subgroup_5_or;Subgroup_5_fa;Subgroup_5_ge; otu0013 1 0.02 13 0.32 Bacteria;Acidobacteria;Subgroup_6;Subgroup_6_or;Subgroup_6_fa;Subgroup_6_ge; otu0014 0 0 1 0.02 Bacteria;Acidobacteria;Subgroup_6;Subgroup_6_un;Subgroup_6_un;Subgroup_6_un; otu0015 8 0.13 30 0.73 Bacteria;Acidobacteria;Subgroup_9;Subgroup_9_or;Subgroup_9_fa;Subgroup_9_ge; -

Prevalence of Ureaplasma Urealyticum, Mycoplasma Hominis and Chlamydia Trachomatis in Patients with Uncomplicated Recurrent Urin
Nephrology and Renal Diseases Research Article ISSN: 2399-908X Prevalence of Ureaplasma urealyticum, Mycoplasma hominis and Chlamydia trachomatis in patients with uncomplicated recurrent urinary tract infections Jadranka Vlasic-Matas1*, Hrvoje Raos2, Marijana Vuckovic2, Stjepan Radic2 and Vesna Capkun3 1Polyclinic Nephrology Department, Split, Croatia 2School of Medicine, University of Split, Split, Croatia 3Department of Nuclear Medicine, Split University Hospital Center, Split, Croatia Abstract Aim: To assess the prevalence of Ureaplasma urealyticum, Mycoplasma hominis and Chlamydia trachomatis in patients with chronic urinary tract infections (UTIs) and its correlation with leukocyturia and symptoms. Methods: The study included 220 patients (130 women and 90 men) presenting with chronic voiding symptoms and sterile leukocyturia. Urine, urethral swabs and cervical swabs (for women patients) were taken to determine the presence of these pathogens. Patients were treated by tetracycline and followed up three and six months after initial therapy. Results: In 186 (85%) out of 220 patients, U. urealyticum was found, while C. trachomatis was present in 34 patients (15%). In majority of female patients (112 out of 130; 86%) U. urealyticum was found. In addition to ureaplasma, in eight patients M. hominis was found. C. trachomatis was identified in 18 female patients (14%). In 74 out of 90 (82%) male patients U. urealyticum was detected while in six of them M. hominis was also found. C. trachomatis was identified in 16 male patients (18%). U. urealyticum was significantly related to leukocyturia, as opposed to C. trachomatis (p<0,001). Women had more frequent symptomatology (p = 0,015) and higer leukocyturia (p<0.001). Conclusion: Leukocyturia is more common find in U.