Identification of Key Genes and Associated Pathways In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
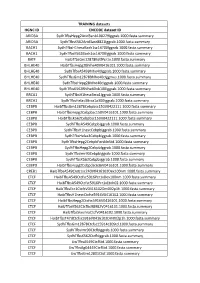
TRAINING Datasets HGNC ID ENCODE Dataset ID ARID3A
TRAINING datasets HGNC ID ENCODE dataset ID ARID3A SydhT+sHepg2Arid3anb100279Iggrab.1000.fasta.summary ARID3A SydhT+sK562Arid3asC8821Iggrab.1000.fasta.summary BACH1 SydhT+sH1hesCBaCh1sC14700Iggrab.1000.fasta.summary BACH1 SydhT+sK562BaCh1sC14700Iggrab.1000.fasta.summary BATF HaibT+sGm12878BaJPCr1x.1000.fasta.summary BHLHE40 HaibT+sHepg2Bhlhe40V0416101.1000.fasta.summary BHLHE40 SydhT+sA549Bhlhe40Iggrab.1000.fasta.summary BHLHE40 SydhT+sGm12878Bhlhe40CIggmus.1000.fasta.summary BHLHE40 SydhT+sHepg2Bhlhe40CIggrab.1000.fasta.summary BHLHE40 SydhT+sK562Bhlhe40nb100Iggrab.1000.fasta.summary BRCA1 SydhT+sH1hesCBrCa1Iggrab.1000.fasta.summary BRCA1 SydhT+sHelas3BrCa1a300Iggrab.1000.fasta.summary CEBPB HaibT+sGm12878CebpbsC150V0422111.1000.fasta.summary CEBPB HaibT+sHepg2CebpbsC150V0416101.1000.fasta.summary CEBPB HaibT+sK562CebpbsC150V0422111.1000.fasta.summary CEBPB SydhT+sA549CebpbIggrab.1000.fasta.summary CEBPB SydhT+sH1hesCCebpbIggrab.1000.fasta.summary CEBPB SydhT+sHelas3CebpbIggrab.1000.fasta.summary CEBPB SydhT+sHepg2CebpbForsklnStd.1000.fasta.summary CEBPB SydhT+sHepg2CebpbIggrab.1000.fasta.summary CEBPB SydhT+sImr90CebpbIggrab.1000.fasta.summary CEBPB SydhT+sK562CebpbIggrab.1000.fasta.summary CEBPD HaibT+sHepg2CebpdsC636V0416101.1000.fasta.summary CREB1 HaibT+sA549Creb1sC240V0416102Dex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xDex100nm.1000.fasta.summary CTCF HaibT+sA549CtCfsC5916PCr1xEtoh02.1000.fasta.summary CTCF HaibT+sECC1CtCfCV0416102Dm002p1h.1000.fasta.summary CTCF HaibT+sH1hesCCtCfsC5916V0416102.1000.fasta.summary -

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

Modes of Interaction of KMT2 Histone H3 Lysine 4 Methyltransferase/COMPASS Complexes with Chromatin
cells Review Modes of Interaction of KMT2 Histone H3 Lysine 4 Methyltransferase/COMPASS Complexes with Chromatin Agnieszka Bochy ´nska,Juliane Lüscher-Firzlaff and Bernhard Lüscher * ID Institute of Biochemistry and Molecular Biology, Medical School, RWTH Aachen University, Pauwelsstrasse 30, 52057 Aachen, Germany; [email protected] (A.B.); jluescher-fi[email protected] (J.L.-F.) * Correspondence: [email protected]; Tel.: +49-241-8088850; Fax: +49-241-8082427 Received: 18 January 2018; Accepted: 27 February 2018; Published: 2 March 2018 Abstract: Regulation of gene expression is achieved by sequence-specific transcriptional regulators, which convey the information that is contained in the sequence of DNA into RNA polymerase activity. This is achieved by the recruitment of transcriptional co-factors. One of the consequences of co-factor recruitment is the control of specific properties of nucleosomes, the basic units of chromatin, and their protein components, the core histones. The main principles are to regulate the position and the characteristics of nucleosomes. The latter includes modulating the composition of core histones and their variants that are integrated into nucleosomes, and the post-translational modification of these histones referred to as histone marks. One of these marks is the methylation of lysine 4 of the core histone H3 (H3K4). While mono-methylation of H3K4 (H3K4me1) is located preferentially at active enhancers, tri-methylation (H3K4me3) is a mark found at open and potentially active promoters. Thus, H3K4 methylation is typically associated with gene transcription. The class 2 lysine methyltransferases (KMTs) are the main enzymes that methylate H3K4. KMT2 enzymes function in complexes that contain a necessary core complex composed of WDR5, RBBP5, ASH2L, and DPY30, the so-called WRAD complex. -

Investigating the Genetic Basis of Cisplatin-Induced Ototoxicity in Adult South African Patients
--------------------------------------------------------------------------- Investigating the genetic basis of cisplatin-induced ototoxicity in adult South African patients --------------------------------------------------------------------------- by Timothy Francis Spracklen SPRTIM002 SUBMITTED TO THE UNIVERSITY OF CAPE TOWN In fulfilment of the requirements for the degree MSc(Med) Faculty of Health Sciences UNIVERSITY OF CAPE TOWN University18 December of Cape 2015 Town Supervisor: Prof. Rajkumar S Ramesar Co-supervisor: Ms A Alvera Vorster Division of Human Genetics, Department of Pathology, University of Cape Town 1 The copyright of this thesis vests in the author. No quotation from it or information derived from it is to be published without full acknowledgement of the source. The thesis is to be used for private study or non- commercial research purposes only. Published by the University of Cape Town (UCT) in terms of the non-exclusive license granted to UCT by the author. University of Cape Town Declaration I, Timothy Spracklen, hereby declare that the work on which this dissertation/thesis is based is my original work (except where acknowledgements indicate otherwise) and that neither the whole work nor any part of it has been, is being, or is to be submitted for another degree in this or any other university. I empower the university to reproduce for the purpose of research either the whole or any portion of the contents in any manner whatsoever. Signature: Date: 18 December 2015 ' 2 Contents Abbreviations ………………………………………………………………………………….. 1 List of figures …………………………………………………………………………………... 6 List of tables ………………………………………………………………………………….... 7 Abstract ………………………………………………………………………………………… 10 1. Introduction …………………………………………………………………………………. 11 1.1 Cancer …………………………………………………………………………….. 11 1.2 Adverse drug reactions ………………………………………………………….. 12 1.3 Cisplatin …………………………………………………………………………… 12 1.3.1 Cisplatin’s mechanism of action ……………………………………………… 13 1.3.2 Adverse reactions to cisplatin therapy ………………………………………. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Comparative Transcriptome Analysis of Embryo Invasion in the Mink Uterus
Placenta 75 (2019) 16–22 Contents lists available at ScienceDirect Placenta journal homepage: www.elsevier.com/locate/placenta Comparative transcriptome analysis of embryo invasion in the mink uterus T ∗ Xinyan Caoa,b, , Chao Xua,b, Yufei Zhanga,b, Haijun Weia,b, Yong Liuc, Junguo Caoa,b, Weigang Zhaoa,b, Kun Baoa,b, Qiong Wua,b a Institute of Special Animal and Plant Sciences, Chinese Academy of Agricultural Sciences, Changchun, China b State Key Laboratory for Molecular Biology of Special Economic Animal and Plant Science, Chinese Academy of Agricultural Sciences, Changchun, China c Key Laboratory of Embryo Development and Reproductive Regulation of Anhui Province, College of Biological and Food Engineering, Fuyang Teachers College, Fuyang, China ABSTRACT Introduction: In mink, as many as 65% of embryos die during gestation. The causes and the mechanisms of embryonic mortality remain unclear. The purpose of our study was to examine global gene expression changes during embryo invasion in mink, and thereby to identify potential signaling pathways involved in implantation failure and early pregnancy loss. Methods: Illumina's next-generation sequencing technology (RNA-Seq) was used to analyze the differentially expressed genes (DEGs) in implantation (IMs) and inter- implantation sites (inter-IMs) of uterine tissue. Results: We identified a total of 606 DEGs, including 420 up- and 186 down-regulated genes in IMs compared to inter-IMs. Gene annotation analysis indicated multiple biological pathways to be significantly enriched for DEGs, including immune response, ECM complex, cytokine activity, chemokine activity andprotein binding. The KEGG pathway including cytokine-cytokine receptor interaction, Jak-STAT, TNF and the chemokine signaling pathway were the most enriched. -

Supplemental Table 1. Complete Gene Lists and GO Terms from Figure 3C
Supplemental Table 1. Complete gene lists and GO terms from Figure 3C. Path 1 Genes: RP11-34P13.15, RP4-758J18.10, VWA1, CHD5, AZIN2, FOXO6, RP11-403I13.8, ARHGAP30, RGS4, LRRN2, RASSF5, SERTAD4, GJC2, RHOU, REEP1, FOXI3, SH3RF3, COL4A4, ZDHHC23, FGFR3, PPP2R2C, CTD-2031P19.4, RNF182, GRM4, PRR15, DGKI, CHMP4C, CALB1, SPAG1, KLF4, ENG, RET, GDF10, ADAMTS14, SPOCK2, MBL1P, ADAM8, LRP4-AS1, CARNS1, DGAT2, CRYAB, AP000783.1, OPCML, PLEKHG6, GDF3, EMP1, RASSF9, FAM101A, STON2, GREM1, ACTC1, CORO2B, FURIN, WFIKKN1, BAIAP3, TMC5, HS3ST4, ZFHX3, NLRP1, RASD1, CACNG4, EMILIN2, L3MBTL4, KLHL14, HMSD, RP11-849I19.1, SALL3, GADD45B, KANK3, CTC- 526N19.1, ZNF888, MMP9, BMP7, PIK3IP1, MCHR1, SYTL5, CAMK2N1, PINK1, ID3, PTPRU, MANEAL, MCOLN3, LRRC8C, NTNG1, KCNC4, RP11, 430C7.5, C1orf95, ID2-AS1, ID2, GDF7, KCNG3, RGPD8, PSD4, CCDC74B, BMPR2, KAT2B, LINC00693, ZNF654, FILIP1L, SH3TC1, CPEB2, NPFFR2, TRPC3, RP11-752L20.3, FAM198B, TLL1, CDH9, PDZD2, CHSY3, GALNT10, FOXQ1, ATXN1, ID4, COL11A2, CNR1, GTF2IP4, FZD1, PAX5, RP11-35N6.1, UNC5B, NKX1-2, FAM196A, EBF3, PRRG4, LRP4, SYT7, PLBD1, GRASP, ALX1, HIP1R, LPAR6, SLITRK6, C16orf89, RP11-491F9.1, MMP2, B3GNT9, NXPH3, TNRC6C-AS1, LDLRAD4, NOL4, SMAD7, HCN2, PDE4A, KANK2, SAMD1, EXOC3L2, IL11, EMILIN3, KCNB1, DOK5, EEF1A2, A4GALT, ADGRG2, ELF4, ABCD1 Term Count % PValue Genes regulation of pathway-restricted GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, SMAD protein phosphorylation 9 6.34 1.31E-08 ENG pathway-restricted SMAD protein GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, phosphorylation -

A Functional Study of ADAMTS7 Gene Variants
A functional study of ADAMTS7 gene variants Xiangyuan Pu A thesis submitted to the University of London for the degree of Doctor of Philosophy Centre for Clinical Pharmacology William Harvey Research Institute Barts and the London School of Medicine and Dentistry Queen Mary, University of London March, 2014 I dedicate this thesis to my parents and my fiancée. Without their endless love, support and encouragement, none of my achievements would be possible. 2 Abstract Background: Recent studies have revealed an association between genetic variants at the ADAMTS7 (a disintegrin-like and metalloprotease with thrombospondin type 1 motif, 7) locus and susceptibility to coronary artery disease (CAD). ADAMTS-7 has been reported to facilitate vascular smooth muscle cell (VSMC) migration and promote neointima formation. We sought to study the functional mechanisms underlying this relationship and to further investigate the role of ADAMTS-7 in atherosclerosis. Methods and Results: In vitro assays showed that the CAD-associated non-synonymous single nucleotide polymorphism rs3825807, which results in a serine to proline (Ser-to- Pro) substitution at residue 214 in the ADAMTS-7 pro-domain, affected ADAMTS-7 pro- domain cleavage. Immunohistochemical analyses showed that ADAMTS-7 localised to vascular smooth muscle cells (VSMCs) and endothelial cells (ECs) in human coronary and carotid atherosclerotic plaques. Cell migration assays demonstrated that VSMCs and ECs from individuals who were homozygous for the adenine (A) allele (encoding the Ser214 isoform) had increased migratory ability compared with cells from individuals who were homozygous for the G allele (encoding the Pro214 isoform). Western blot analyses revealed that media conditioned by VSMCs of the A/A genotype contained more cleaved ADAMTS-7 pro-domain and more of the cleaved form of thrombospondin-5 (TSP-5, an ADAMTS-7 substrate that had been shown to be produced by VSMCs and inhibit VSMC migration). -

Transcriptome-Wide Profiling of Cerebral Cavernous Malformations
www.nature.com/scientificreports OPEN Transcriptome-wide Profling of Cerebral Cavernous Malformations Patients Reveal Important Long noncoding RNA molecular signatures Santhilal Subhash 2,8, Norman Kalmbach3, Florian Wegner4, Susanne Petri4, Torsten Glomb5, Oliver Dittrich-Breiholz5, Caiquan Huang1, Kiran Kumar Bali6, Wolfram S. Kunz7, Amir Samii1, Helmut Bertalanfy1, Chandrasekhar Kanduri2* & Souvik Kar1,8* Cerebral cavernous malformations (CCMs) are low-fow vascular malformations in the brain associated with recurrent hemorrhage and seizures. The current treatment of CCMs relies solely on surgical intervention. Henceforth, alternative non-invasive therapies are urgently needed to help prevent subsequent hemorrhagic episodes. Long non-coding RNAs (lncRNAs) belong to the class of non-coding RNAs and are known to regulate gene transcription and involved in chromatin remodeling via various mechanism. Despite accumulating evidence demonstrating the role of lncRNAs in cerebrovascular disorders, their identifcation in CCMs pathology remains unknown. The objective of the current study was to identify lncRNAs associated with CCMs pathogenesis using patient cohorts having 10 CCM patients and 4 controls from brain. Executing next generation sequencing, we performed whole transcriptome sequencing (RNA-seq) analysis and identifed 1,967 lncRNAs and 4,928 protein coding genes (PCGs) to be diferentially expressed in CCMs patients. Among these, we selected top 6 diferentially expressed lncRNAs each having signifcant correlative expression with more than 100 diferentially expressed PCGs. The diferential expression status of the top lncRNAs, SMIM25 and LBX2-AS1 in CCMs was further confrmed by qRT-PCR analysis. Additionally, gene set enrichment analysis of correlated PCGs revealed critical pathways related to vascular signaling and important biological processes relevant to CCMs pathophysiology. -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Investigation of the Underlying Hub Genes and Molexular Pathogensis in Gastric Cancer by Integrated Bioinformatic Analyses
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Investigation of the underlying hub genes and molexular pathogensis in gastric cancer by integrated bioinformatic analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The high mortality rate of gastric cancer (GC) is in part due to the absence of initial disclosure of its biomarkers. The recognition of important genes associated in GC is therefore recommended to advance clinical prognosis, diagnosis and and treatment outcomes. The current investigation used the microarray dataset GSE113255 RNA seq data from the Gene Expression Omnibus database to diagnose differentially expressed genes (DEGs). Pathway and gene ontology enrichment analyses were performed, and a proteinprotein interaction network, modules, target genes - miRNA regulatory network and target genes - TF regulatory network were constructed and analyzed. Finally, validation of hub genes was performed. The 1008 DEGs identified consisted of 505 up regulated genes and 503 down regulated genes. -

Supplementary Material
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Page 1 / 45 SUPPLEMENTARY MATERIAL Appendix A1: Neuropsychological protocol. Appendix A2: Description of the four cases at the transitional stage. Table A1: Clinical status and center proportion in each batch. Table A2: Complete output from EdgeR. Table A3: List of the putative target genes. Table A4: Complete output from DIANA-miRPath v.3. Table A5: Comparison of studies investigating miRNAs from brain samples. Figure A1: Stratified nested cross-validation. Figure A2: Expression heatmap of miRNA signature. Figure A3: Bootstrapped ROC AUC scores. Figure A4: ROC AUC scores with 100 different fold splits. Figure A5: Presymptomatic subjects probability scores. Figure A6: Heatmap of the level of enrichment in KEGG pathways. Kmetzsch V, et al. J Neurol Neurosurg Psychiatry 2021; 92:485–493. doi: 10.1136/jnnp-2020-324647 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Appendix A1. Neuropsychological protocol The PREV-DEMALS cognitive evaluation included standardized neuropsychological tests to investigate all cognitive domains, and in particular frontal lobe functions. The scores were provided previously (Bertrand et al., 2018). Briefly, global cognitive efficiency was evaluated by means of Mini-Mental State Examination (MMSE) and Mattis Dementia Rating Scale (MDRS). Frontal executive functions were assessed with Frontal Assessment Battery (FAB), forward and backward digit spans, Trail Making Test part A and B (TMT-A and TMT-B), Wisconsin Card Sorting Test (WCST), and Symbol-Digit Modalities test.