Immunologic Risk Prediction Model for Kidney Graft Function
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pheasant Season Slow to the Last Veteran, Is Dead Red Cross Drive
Nebrask~ st~te Historical ooctety \. I THE . r i' '. .• '•.' .... '. ..~ . I r RED CROSS "The Paper \Vith The Pictures" "Read by 3~000 Families Every \Veeh RED CROSS Established April, 1882 THE ORO QUIZ, ORO, NEBRASKA \VEONESDAY, NOVEMBER 5. 1941 Vol. 59 No, 32 New Chevrolet Brings Grin to \Vinner's Face Four Young People Escaped Death in This \Vreck I Chanticleers Take John M. Lindsay, ~ 11942 Fair to Be LOUll City 34 to 0 Last Civil War Held on Grounds in Fast Grid Ganle Veteran, Is Dead Late in August On Defensive First Frame, Burwell Man Heeds Call at Clement, McGinnis, Dale and Ord Comes Back strong to Age of 93; Served Nineteen Misko, Reelected Officers; Continue Title March, Months in Iowa Infantry, Pearson New Director. On the detensive throughout the first quarter of their game with Burwell- (Spedal)-The last of Decision to hold the 1942 Valley Loup ·Cily at 13ussell park field Fri the Boys in Blue answered the county fair on the fair grounds in day evening, the OrO. Chanticleers final' tans and passed from the its entirely rather than to have en- came back strong and won 34 to o. earthly scene when John M. Lind- tertalmuent features "Oil the streets • .Apparently the Chanticleers were say, 93, 'who' is 'belleH;d to have and exhibits Oil the grounds was victims of over-confidenee Iroiu been the last surviving veteran of made on Monday at the annual their lopsided win over Ravenna as the Civil War. in thls part of the meeting of stockholders held in the the ganie against Loup City began state, died in the home of his niece, district court room here, It was and early in the period they found Who wouldn't grin it he suddenly learned that he had won a new Mrs. -

LCSH Section K
K., Rupert (Fictitious character) Motion of K stars in line of sight Ka-đai language USE Rupert (Fictitious character : Laporte) Radial velocity of K stars USE Kadai languages K-4 PRR 1361 (Steam locomotive) — Orbits Ka’do Herdé language USE 1361 K4 (Steam locomotive) UF Galactic orbits of K stars USE Herdé language K-9 (Fictitious character) (Not Subd Geog) K stars—Galactic orbits Ka’do Pévé language UF K-Nine (Fictitious character) BT Orbits USE Pévé language K9 (Fictitious character) — Radial velocity Ka Dwo (Asian people) K 37 (Military aircraft) USE K stars—Motion in line of sight USE Kadu (Asian people) USE Junkers K 37 (Military aircraft) — Spectra Ka-Ga-Nga script (May Subd Geog) K 98 k (Rifle) K Street (Sacramento, Calif.) UF Script, Ka-Ga-Nga USE Mauser K98k rifle This heading is not valid for use as a geographic BT Inscriptions, Malayan K.A.L. Flight 007 Incident, 1983 subdivision. Ka-houk (Wash.) USE Korean Air Lines Incident, 1983 BT Streets—California USE Ozette Lake (Wash.) K.A. Lind Honorary Award K-T boundary Ka Iwi National Scenic Shoreline (Hawaii) USE Moderna museets vänners skulpturpris USE Cretaceous-Paleogene boundary UF Ka Iwi Scenic Shoreline Park (Hawaii) K.A. Linds hederspris K-T Extinction Ka Iwi Shoreline (Hawaii) USE Moderna museets vänners skulpturpris USE Cretaceous-Paleogene Extinction BT National parks and reserves—Hawaii K-ABC (Intelligence test) K-T Mass Extinction Ka Iwi Scenic Shoreline Park (Hawaii) USE Kaufman Assessment Battery for Children USE Cretaceous-Paleogene Extinction USE Ka Iwi National Scenic Shoreline (Hawaii) K-B Bridge (Palau) K-TEA (Achievement test) Ka Iwi Shoreline (Hawaii) USE Koro-Babeldaod Bridge (Palau) USE Kaufman Test of Educational Achievement USE Ka Iwi National Scenic Shoreline (Hawaii) K-BIT (Intelligence test) K-theory Ka-ju-ken-bo USE Kaufman Brief Intelligence Test [QA612.33] USE Kajukenbo K. -

Spring 2020 Virtual Commencement Exercises Click Here to View Ceremonies
SOUTHERN NEW HAMPSHIRE UNIVERSITY SPRING 2020 VIRTUAL COMMENCEMENT EXERCISES CLICK HERE TO VIEW CEREMONIES SATURDAY, MAY 8, 12 PM ET 2021 TABLE OF CONTENTS CONFERRAL GRADUATE AND UNDERGRADUATE DEGREES ........................................ 1 SNHU Honor Societies Honor Society Listing .................................................................................................. 3 Presentation of Degree Candidates COLLEGE FOR AMERICA .............................................................................................. 6 BUSINESS PROGRAMS ................................................................................................ 15 COUNSELING PROGRAMS ........................................................................................... 57 EDUCATION PROGRAMS ............................................................................................ 59 HEALTHCARE PROGRAMS .......................................................................................... 62 LIBERAL ARTS PROGRAMS .........................................................................................70 NURSING PROGRAMS .................................................................................................92 SOCIAL SCIENCE PROGRAMS ..................................................................................... 99 SCIENCE, TECHNOLOGY, ENGINEERING AND MATH (STEM) PROGRAMS ................... 119 Post-Ceremony WELCOME FROM THE ALUMNI ASSOCIATION ............................................................ 131 CONFERRAL OF GRADUATE -

British Family Names
cs 25o/ £22, Cornrll IBniwwitg |fta*g BOUGHT WITH THE INCOME FROM THE SAGE ENDOWMENT FUND THE GIFT OF Hcnrti W~ Sage 1891 A.+.xas.Q7- B^llll^_ DATE DUE ,•-? AUG 1 5 1944 !Hak 1 3 1^46 Dec? '47T Jan 5' 48 ft e Univeral, CS2501 .B23 " v Llb«"y Brit mii!Sm?nS,£& ori8'" and m 3 1924 olin 029 805 771 The original of this book is in the Cornell University Library. There are no known copyright restrictions in the United States on the use of the text. http://www.archive.org/details/cu31924029805771 BRITISH FAMILY NAMES. : BRITISH FAMILY NAMES ftbetr ©riain ano fIDeaning, Lists of Scandinavian, Frisian, Anglo-Saxon, and Norman Names. HENRY BARBER, M.D. (Clerk), "*• AUTHOR OF : ' FURNESS AND CARTMEL NOTES,' THE CISTERCIAN ABBEY OF MAULBRONN,' ( SOME QUEER NAMES,' ' THE SHRINE OF ST. BONIFACE AT FULDA,' 'POPULAR AMUSEMENTS IN GERMANY,' ETC. ' "What's in a name ? —Romeo and yuliet. ' I believe now, there is some secret power and virtue in a name.' Burton's Anatomy ofMelancholy. LONDON ELLIOT STOCK, 62, PATERNOSTER ROW, E.C. 1894. 4136 CONTENTS. Preface - vii Books Consulted - ix Introduction i British Surnames - 3 nicknames 7 clan or tribal names 8 place-names - ii official names 12 trade names 12 christian names 1 foreign names 1 foundling names 1 Lists of Ancient Patronymics : old norse personal names 1 frisian personal and family names 3 names of persons entered in domesday book as HOLDING LANDS temp. KING ED. CONFR. 37 names of tenants in chief in domesday book 5 names of under-tenants of lands at the time of the domesday survey 56 Norman Names 66 Alphabetical List of British Surnames 78 Appendix 233 PREFACE. -

Smart Hybridity
SMART HYBRIDITY SMART SMART HYBRIDITY Potentials and Challenges of New Governance Arrangements Koppenjan, Karré, Termeer (Eds.) Koppenjan, Karré, Termeer Joop Koppenjan Philip Marcel Karré Katrien Termeer (Eds.) In our current society, governments face complex societal issues that cannot be tackled through traditional governance arrangements. Therefore, governments increasingly come up with smart hybrid arrange ments that transcend the boundaries of policy domains and jurisdictions, combine governance mechanisms (state, market, net- works and self-governance), and foster new forms of collaboration. HYBRIDITY SMART This book provides an overview of what smart hybridity entails and of its potentials and challenges. It includes empirical analyses of hybrid arrangements in five policy domains, and reflections upon these studies by inter nationally renowned governance scholars. They show that the smartness of the new hybrid arrangements does not lie in realizing quick fixes, but in partici pants’ capacities to learn, adapt and arrive at sustainable and legitimate solutions that balance various public values. ‘Hybridity increasingly defines the governance, structure and opera- tion of contemporary social institutions. Yet, surprisingly, there is limited insight available as to the decision-making and effectiveness of the various compositions that hybrid governance modes may take. This edited volume addresses this shortfall by way of a series of applied case studies that serve to unpack the various mixes of gover nance arrangements, their features and operating processes of hybrid arrangements across several fields. It will be particularly useful for those charged with designing, implementing and evaluat- ing the ‘smarter’ hybrid governance arrangements required for our (Eds.) Koppenjan, Karré, Termeer in creasingly complex and challenging contexts.’ Professor Robyn Keast, Southern Cross University, Gold Coast, Australia. -

NOTICE to Consumers, and Former Consumers, of Poudre Valley REA for Unclaimed Funds REFER to THIS LIST to SEE IF YOU ARE OWED MONEY from POUDRE VALLEY REA
NOTICE To Consumers, and Former Consumers, of Poudre Valley REA for Unclaimed Funds REFER TO THIS LIST TO SEE IF YOU ARE OWED MONEY FROM POUDRE VALLEY REA Each year the Poudre Valley REA Board of Directors reviews the Association’s financial health and determines how If you were a consumer of Poudre Valley REA much, if any, retained capital should be returned to our during any of the years 1993, 2004, and/or members. Last year the board of directors approved the 2018, please look for your name in this notice. retirement of $3.2 million in allocated capital credits from 1993, 2004, and/or 2018. Consumers who received service If you see a name you recognize contact us at: from Poudre Valley REA in those years, and accumulated Poudre Valley REA retired credits of $10 or more, received a check in the mail PO Box 272550 or a bill credit last September. PVREA was unable to return $125,918 to 4,248 of our members because of a bad address Ft. Collins, CO 80527-2550 on file or the member simply did not cash the check. If your name is on the list, give us a call so we can determine if you 800-432-1012 have money coming to you. 1ST FREE WILL BAPTIST AFFINITY HOMES AMBRIZ, MICHAEL APPLE, TRACY P 360 NETWORKS USA INC AGEE, RICHARD E AMEN, CARL ARAGON, SYLVIA L 4 J L CORPORATION AHLBRANDT, DEBORAH K AMES, CHARLES T ARAKAWA, NORMAN D A - G SOD FARMS INC AHLSCHWEDE, DELVIN J AMES, JEREMY ARBANAS, JERRY A & J ANTIQUES & COLLECT- AINSWORTH, THOMAS AMOS, R D ARBINI, SUSAN K ABLES AIRHART, ROBERT L AMUNDSEN, MICHAEL ARBO, ANNMARIE A B F FREIGHT SYSTEM ALBERT, -

Last Name First Name Page Volume SAALI JUDY ANN 515 4
Last Name First Name Page Volume SAALI JUDY ANN 515 4 SAATHOFF DONALD JOHN 244 14 SAATHOFF DALE FREDERICK 98 19 SAATHOFF MIRANDA LEIGH 266 26 SAATHOFF MONICA LYNN 135 28 SAATHOFF STEVEN GUERRA 15 36 SABADO CLAUDIA ANN 176 13 SABALA RICHARD MANUEL JR 232 8 SABATKA ANNIE 235 2 SABATKA JOE 302 3 SABLE FRED ALLEN 295 6 SABY MARY ELLEN 106 5 SACHARIASON PETER JOEL 165 28 SACHER WILLIAM HART 23 21 SACHWITZ SHANE ROBERT COLE 83 40 SACIA TERRY ALAN 272 22 SACK JACQUELINE PATRICE 192 36 SACKETT DOROTHY D. 324 2 SACKETT KENNETH LEROY 225 5 SADLER JAMES ARTHUR 4 16 SADLER MONICA ELAINE 198 39 SAENZ GUADALUPE CHAVEZ 145 12 SAENZ-ZUBIA TANIA CONCEPCION 201 42 SAFFEL JOSEPH CLEM 489 5 SAFFELL SHERILYN GAYLE 206 41 SAFFORD REASA DAWN 65 40 SAFORA EDISON 101 39 SAGE GLENDA MARIE 611 5 SAGE NICOLE LYNN 63 39 SAGER ANNA DREW JACE 109 4 SAGER VANESSA MARIE 48 14 SAGERT ALAN CURTTISS 35 7 SAHL JENNIFER DELYNN 42 24 SAILER KATHY EVELYN 251 14 SAINDON JAMES LEE 212 8 SAINSBURY RICHARD LYNN 228 9 SAINSBURY MARGARET ROSE 3 15 SAIZAN DONNA KAY 150 24 SAIZAN ANGELA MICHELLE 42 26 SAKIS PAUL EMIL 412 6 SALAS PEREA IVAN 5 38 SALAZAR ESTHER IDA 242 6 SALAZAR JAIMEE RAYE 31 32 SALAZAR SIERRA SUMMER 141 42 SALAZER MANUEL EUGENE 26 19 SALDIVAR-FLORES MARIA GUADALUPE 157 40 SALEY DOUGLAS JOHN 186 10 SALEY GRACE CATHERINE 217 13 SALIMINEN DOROTHY FAYE 12 16 SALIMINEN LAWRENCE WESLEY 12 16 SALINAS AMANDA MARIE 17 28 SALISBURY BRYAN STUART 201 17 SALISBURY ALEXIA NMN 95 27 SALISBURY BRYAN STUART 116 40 SALMON MALISSA MARIE 223 20 SALMON CRYSTAL MARIE 177 37 SALMONSON DAVID -
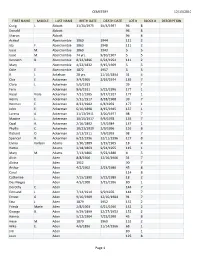
12/13/2010 CEMETERY Page 1 FIRST NAME MIDDLE LAST NAME
CEMETERY 12/13/2010 FIRST NAME MIDDLE LAST NAME BIRTH DATE DEATH DATE LOT # BLOCK # DESCRIPTION Craig L. Abbott 11/20/1975 10/4/1997 96 8 Donald Abbott 96 8 Sharon Abbott 96 8 Acricol Abercrombie 1860 1944 111 2 Ida F. Abercrombie 1863 1948 111 2 Isaac M. Abercrombie 1869 1943 5 5 Issac M. Abercrombie 74 yrs. 9/10/1907 5 5 Kenneth B. Abercrombie 8/24/1896 6/24/1954 111 2 Mary Abercrombie 4/22/1832 9/15/1909 5 5 Odie E. Abercrombie 1872 1957 5 5 R. J. Ackelson 28 yrs. 11/10/1894 31 4 Chis E. Ackerman 3/4/1905 2/19/1974 135 7 Clara L. Ackerman 5/6/1913 39 7 Fern Ackerman 8/6/1911 5/22/1996 177 1 Hazel Viola Ackerman 7/31/1905 9/17/1927 177 1 Henry E. Ackerman 5/31/1917 8/18/1968 39 7 Herman E. Ackerman 8/22/1902 6/4/1959 177 1 John E. Ackerman 6/10/1898 4/15/1945 137 1 Lorena A. Ackerman 11/13/1911 3/20/1977 98 7 Maxine L. Ackerman 10/15/1917 9/6/1978 135 7 Opal A. Ackerman 2/16/1892 7/3/1984 137 1 Phyllis C. Ackerman 10/21/1939 2/9/1996 126 8 Richard O. Ackerman 2/13/1911 7/6/1993 98 7 Spencer R. Ackerman 6/22/1996 12/11/1996 127 8 Elvina Karlson Adams 1/30/1889 1/19/1965 18 4 Hattie Adams 2/18/1863 9/24/1955 115 1 Mary M. -

Annexure 1B 18416
Annexure 1 B List of taxpayers allotted to State having turnover of more than or equal to 1.5 Crore Sl.No Taxpayers Name GSTIN 1 BROTHERS OF ST.GABRIEL EDUCATION SOCIETY 36AAAAB0175C1ZE 2 BALAJI BEEDI PRODUCERS PRODUCTIVE INDUSTRIAL COOPERATIVE SOCIETY LIMITED 36AAAAB7475M1ZC 3 CENTRAL POWER RESEARCH INSTITUTE 36AAAAC0268P1ZK 4 CO OPERATIVE ELECTRIC SUPPLY SOCIETY LTD 36AAAAC0346G1Z8 5 CENTRE FOR MATERIALS FOR ELECTRONIC TECHNOLOGY 36AAAAC0801E1ZK 6 CYBER SPAZIO OWNERS WELFARE ASSOCIATION 36AAAAC5706G1Z2 7 DHANALAXMI DHANYA VITHANA RAITHU PARASPARA SAHAKARA PARIMITHA SANGHAM 36AAAAD2220N1ZZ 8 DSRB ASSOCIATES 36AAAAD7272Q1Z7 9 D S R EDUCATIONAL SOCIETY 36AAAAD7497D1ZN 10 DIRECTOR SAINIK WELFARE 36AAAAD9115E1Z2 11 GIRIJAN PRIMARY COOPE MARKETING SOCIETY LIMITED ADILABAD 36AAAAG4299E1ZO 12 GIRIJAN PRIMARY CO OP MARKETING SOCIETY LTD UTNOOR 36AAAAG4426D1Z5 13 GIRIJANA PRIMARY CO-OPERATIVE MARKETING SOCIETY LIMITED VENKATAPURAM 36AAAAG5461E1ZY 14 GANGA HITECH CITY 2 SOCIETY 36AAAAG6290R1Z2 15 GSK - VISHWA (JV) 36AAAAG8669E1ZI 16 HASSAN CO OPERATIVE MILK PRODUCERS SOCIETIES UNION LTD 36AAAAH0229B1ZF 17 HCC SEW MEIL JOINT VENTURE 36AAAAH3286Q1Z5 18 INDIAN FARMERS FERTILISER COOPERATIVE LIMITED 36AAAAI0050M1ZW 19 INDU FORTUNE FIELDS GARDENIA APARTMENT OWNERS ASSOCIATION 36AAAAI4338L1ZJ 20 INDUR INTIDEEPAM MUTUAL AIDED CO-OP THRIFT/CREDIT SOC FEDERATION LIMITED 36AAAAI5080P1ZA 21 INSURANCE INFORMATION BUREAU OF INDIA 36AAAAI6771M1Z8 22 INSTITUTE OF DEFENCE SCIENTISTS AND TECHNOLOGISTS 36AAAAI7233A1Z6 23 KARNATAKA CO-OPERATIVE MILK PRODUCER\S FEDERATION -

Download the Every-Name Index to the 1890 St. Louis County
St. Louis County Watchman-Advocate [email protected] Special Collecitons Department Every-name index, 1890 www.slcl.org St. Louis County Library Page 1 Last Name First Name Date Year Page Col Note **igenhanm Mrs 24-Oct 1890 1 a and grandson Abeles Robt 14-Nov 1890 8 e Able Mr 21-Nov 1890 5 d Abrams Emma 23-May 1890 8 b Abrams Geo 21-Feb 1890 8 c Abrams George 10-Oct 1890 8 c Achbach Martin 25-Jul 1890 8 c Achenbach Charles 23-May 1890 8 c Achenbach Charles 6-Jun 1890 8 c Achenbach Charles H 23-May 1890 8 a Achenbach Fred 17-Jan 1890 1 b Achenbach Fred 7-Feb 1890 1 b Achenbach Fred 7-Feb 1890 4 a Achenbach Fred 14-Feb 1890 1 c Achenbach Fred 23-May 1890 4 c Achenbach Fred 23-May 1890 8 a Achenbach Fred 6-Jun 1890 8 a Achenbach Fred 20-Jun 1890 8 a Achenbach Fred 4-Jul 1890 8 a Achenbach Fred 1-Aug 1890 8 a Achenbach Fred 22-Aug 1890 8 a Achenbach Fred 29-Aug 1890 5 b Achenbach Fred 19-Sep 1890 5 c Achenbach Fred 28-Nov 1890 4 c Achenbach Fred 28-Nov 1890 5 a Achenbach Fred 26-Dec 1890 5 c Achenbach Fred 26-Dec 1890 5 d Achenbach Fred 26-Dec 1890 8 a Achenbach Prof 31-Jan 1890 4 a Achenbach Prof 31-Oct 1890 8 c Achenbach Prof 14-Nov 1890 8 b Achenbach Selma 20-Jun 1890 8 a Ackerman Charley 24-Jan 1890 8 c Ackerman F 12-Sep 1890 5 a Ackerman Henry 5-Dec 1890 8 b October 2013 St. -

First Name Middle Name Last Name
First Name Middle Name Last Name Generation First Name Middle Name Last Name Generation Eldie Ray Abel Jane Ellen Hamilton Lacie Nicole Williams Sondra D Perkins Linda Kay Cerniglia James Andrew Posey Jr Shelia Faye Williams LaDean Kaye Marris Bruce Aubrey Benham Matthew Thomas Berry David Paul Smoot Joshua David Kelley Larry Joseph Lansdale Shirley Ann Almerigi Maggie Chenelle Lena John Bard Tyndall Jr Johnnie Mae Brasuell Joe Johnson Margo Goudeau Billy Gene Proctor Jason Turner Holuby Robert Franklyn Herrod Henry Harjo Jr Rebecca Lynn Landsberry Timothy Russell Paulk Richard Hazen Smoot Todd Howitt Morgan Henry Glen Cooper Norman Blane Morton Clonda Evon Rogers Dwight Daniel Sutterfield David Paul Herrod Jessica Dawn Rosalez Clarence Lee Baccus Jr Christopher Franklin Pantle II Michael Lee Parker Quint Henry Miller III Jim Lee Pitman Ralph Marshall Mayberry Larry Dan Langston Tracy Lee Eshleman Leo Wayne Nichols Terry Vezel Butler Sherman Ray Henry Richard Otis Beaver Micco Topley Ispocogee Geraldine Johnson Pulaski Eldon Wayne Marshall Carol Ree Greenwood Julia June Spain Seneca Martin Smith Timothy Eric Yahola Margaret Gay Downing Bruce Allen McAfee Samuel David Marshall Jr Ryan Alexander Hicks Roman Gabriel Rosalez Jr John Richard Yahola Travis Pierce Lewis Thomas Harrell James Daniel Dean Barnett Michael Sherman West Daniel Mcintosh Grant Nancy Ruth Webber Jeremy Dewochee Fultz David Franklin Cox Janet Dee Maylen Alene Barnett Martha May Miller Linda Louise Ogle Sylvanna Georgia Caldwell Albert Jackson Tiger Steven Lee Johnson Desmond -
IL-12-Secreting CD19-Targeted Cord Blood-Derived T Cells for the Immunotherapy of B-Cell Acute Lymphoblastic Leukemia
Leukemia (2015) 29, 415–422 © 2015 Macmillan Publishers Limited All rights reserved 0887-6924/15 www.nature.com/leu ORIGINAL ARTICLE IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia HJ Pegram1, TJ Purdon1, DG van Leeuwen1, KJ Curran2, SA Giralt1,3,4, JN Barker1,3,4 and RJ Brentjens1,5,6 Disease relapse or progression is a major cause of death following umbilical cord blood (UCB) transplantation (UCBT) in patients with high-risk, relapsed or refractory acute lymphoblastic leukemia (ALL). Adoptive transfer of donor-derived T cells modified to express a tumor-targeted chimeric antigen receptor (CAR) may eradicate persistent disease after transplantation. Such therapy has not been available to UCBT recipients, however, due to the low numbers of available UCB T cells and the limited capacity for ex vivo expansion of cytolytic cells. We have developed a novel strategy to expand UCB T cells to clinically relevant numbers in the context of exogenous cytokines. UCB-derived T cells cultured with interleukin (IL)-12 and IL-15 generated 4150-fold expansion with a unique central memory/effector phenotype. Moreover, UCB T cells were modified to both express the CD19-specific CAR, 1928z, and secrete IL-12. 1928z/IL-12 UCB T cells retained a central memory-effector phenotype and had increased antitumor efficacy in vitro. Furthermore, adoptive transfer of 1928z/IL-12 UCB T cells resulted in significantly enhanced survival of CD19+ tumor- bearing SCID-Beige mice. Clinical translation of CAR-modified UCB T cells could augment the graft-versus-leukemia effect after UCBT and thus further improve disease-free survival of transplant patients with B-cell ALL.