Full-Length Envelope Analyzer (FLEA): a Tool for Longitudinal Analysis of Viral Amplicons
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
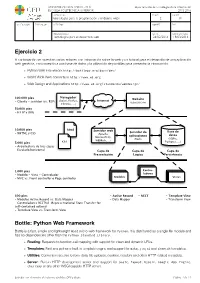
Ejercicio 2 Bottle: Python Web Framework
UNIVERSIDAD SAN PABLO - CEU departamento de tecnologías de la información ESCUELA POLITÉCNICA SUPERIOR 2015-2016 ASIGNATURA CURSO GRUPO tecnologías para la programación y el diseño web i 2 01 CALIFICACION EVALUACION APELLIDOS NOMBRE DNI OBSERVACIONES FECHA FECHA ENTREGA Tecnologías para el desarrollo web 24/02/2016 18/03/2016 Ejercicio 2 A continuación se muestran varios enlaces con información sobre la web y un tutorial para el desarrollo de una aplicación web genérica, con conexión a una base de datos y la utilización de plantillas para presentar la información. ‣ Python Web Framework http://bottlepy.org/docs/dev/ ‣ World Wide Web consortium http://www.w3.org ‣ Web Design and Applications http://www.w3.org/standards/webdesign/ Navegador 100.000 pies Website (Safari, Firefox, Internet - Cliente - servidor (vs. P2P) uspceu.com Chrome, ...) 50.000 pies - HTTP y URIs 10.000 pies html Servidor web Servidor de Base de - XHTML y CSS (Apache, aplicaciones datos Microsoft IIS, (Rack) (SQlite, WEBRick, ...) 5.000 pies css Postgres, ...) - Arquitectura de tres capas - Escalado horizontal Capa de Capa de Capa de Presentación Lógica Persistencia 1.000 pies Contro- - Modelo - Vista - Controlador ladores - MVC vs. Front controller o Page controller Modelos Vistas 500 pies - Active Record - REST - Template View - Modelos Active Record vs. Data Mapper - Data Mapper - Transform View - Controladores RESTful (Representational State Transfer for self-contained actions) - Template View vs. Transform View Bottle: Python Web Framework Bottle is a fast, simple and lightweight WSGI micro web-framework for Python. It is distributed as a single file module and has no dependencies other than the Python Standard Library. -

WEB2PY Enterprise Web Framework (2Nd Edition)
WEB2PY Enterprise Web Framework / 2nd Ed. Massimo Di Pierro Copyright ©2009 by Massimo Di Pierro. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning, or otherwise, except as permitted under Section 107 or 108 of the 1976 United States Copyright Act, without either the prior written permission of the Publisher, or authorization through payment of the appropriate per-copy fee to the Copyright Clearance Center, Inc., 222 Rosewood Drive, Danvers, MA 01923, (978) 750-8400, fax (978) 646-8600, or on the web at www.copyright.com. Requests to the Copyright owner for permission should be addressed to: Massimo Di Pierro School of Computing DePaul University 243 S Wabash Ave Chicago, IL 60604 (USA) Email: [email protected] Limit of Liability/Disclaimer of Warranty: While the publisher and author have used their best efforts in preparing this book, they make no representations or warranties with respect to the accuracy or completeness of the contents of this book and specifically disclaim any implied warranties of merchantability or fitness for a particular purpose. No warranty may be created ore extended by sales representatives or written sales materials. The advice and strategies contained herein may not be suitable for your situation. You should consult with a professional where appropriate. Neither the publisher nor author shall be liable for any loss of profit or any other commercial damages, including but not limited to special, incidental, consequential, or other damages. Library of Congress Cataloging-in-Publication Data: WEB2PY: Enterprise Web Framework Printed in the United States of America. -

A Guide to Brazil's Oil and Oil Derivatives Compliance Requirements
A Guide to Brazil’s Oil and Oil Derivatives Compliance Requirements A Guide to Importing Petroleum Products (Oil and Oil Derivatives) into Brazil 1. Scope 2. General View of the Brazilian Regulatory Framework 3. Regulatory Authorities for Petroleum Products 3.1 ANP’s Technical Regulations 3.2 INMETRO’s Technical Regulations 4. Standards Developing Organizations 4.1 Brazilian Association of Technical Standards (ABNT) 5. Certifications and Testing Bodies 5.1 Certification Laboratories Listed by Inmetro 5.2 Testing Laboratories Listed by Inmetro 6. Government Partners 7. Major Market Entities 1 A Guide to Importing Petroleum Products (Oil and Oil Derivatives) into Brazil 1. Scope This guide addresses all types of petroleum products regulated in Brazil. 2. General Overview of the Brazilian Regulatory Framework Several agencies at the federal level have the authority to issue technical regulations in the particular areas of their competence. Technical regulations are always published in the Official Gazette and are generally based on international standards. All agencies follow similar general procedures to issue technical regulations. They can start the preparation of a technical regulation ex officio or at the request of a third party. If the competent authority deems it necessary, a draft regulation is prepared and published in the Official Gazette, after carrying out an impact assessment of the new technical regulation. Technical regulations take the form of laws, decrees or resolutions. Brazil normally allows a six-month period between the publication of a measure and its entry into force. Public hearings are also a way of promoting the public consultation of the technical regulations. -

For Fuel 1 (BDN) at 550, 1,100 and 1,650 Bar Injection Pressures
City Research Online City, University of London Institutional Repository Citation: Fatmi, Zeeshan (2018). Optical and chemical characterisation of the effects of high-pressure hydrodynamic cavitation on diesel fuel. (Unpublished Doctoral thesis, City, University of London) This is the accepted version of the paper. This version of the publication may differ from the final published version. Permanent repository link: https://openaccess.city.ac.uk/id/eprint/22683/ Link to published version: Copyright: City Research Online aims to make research outputs of City, University of London available to a wider audience. Copyright and Moral Rights remain with the author(s) and/or copyright holders. URLs from City Research Online may be freely distributed and linked to. Reuse: Copies of full items can be used for personal research or study, educational, or not-for-profit purposes without prior permission or charge. Provided that the authors, title and full bibliographic details are credited, a hyperlink and/or URL is given for the original metadata page and the content is not changed in any way. City Research Online: http://openaccess.city.ac.uk/ [email protected] OPTICAL AND CHEMICAL CHARACTERISATION OF THE EFFECTS OF HIGH-PRESSURE HYDRODYNAMIC CAVITATION ON DIESEL FUEL Zeeshan Fatmi This thesis is submitted in fulfilment of the requirements of the degree of Doctor of Philosophy School of Mathematics, Computer Science and Engineering Department of Mechanical Engineering February 2018 i Table of Contents 2.1 Diesel Fuel................................................................................................................... 7 2.1.1 Distillation from Crude Oil and Conversion Processes ....................................... 7 2.1.2 Diesel Fuel Components .................................................................................... 10 2.1.3 Diesel Fuel Properties and Performance Parameters ........................................ -

Introducing Python
Introducing Python Bill Lubanovic Introducing Python by Bill Lubanovic Copyright © 2015 Bill Lubanovic. All rights reserved. Printed in the United States of America. Published by O’Reilly Media, Inc., 1005 Gravenstein Highway North, Sebastopol, CA 95472. O’Reilly books may be purchased for educational, business, or sales promotional use. Online editions are also available for most titles (http://safaribooksonline.com). For more information, contact our corporate/ institutional sales department: 800-998-9938 or [email protected]. Editors: Andy Oram and Allyson MacDonald Indexer: Judy McConville Production Editor: Nicole Shelby Cover Designer: Ellie Volckhausen Copyeditor: Octal Publishing Interior Designer: David Futato Proofreader: Sonia Saruba Illustrator: Rebecca Demarest November 2014: First Edition Revision History for the First Edition: 2014-11-07: First release 2015-02-20: Second release See http://oreilly.com/catalog/errata.csp?isbn=9781449359362 for release details. The O’Reilly logo is a registered trademark of O’Reilly Media, Inc. Introducing Python, the cover image, and related trade dress are trademarks of O’Reilly Media, Inc. Many of the designations used by manufacturers and sellers to distinguish their products are claimed as trademarks. Where those designations appear in this book, and O’Reilly Media, Inc. was aware of a trademark claim, the designations have been printed in caps or initial caps. While the publisher and the author have used good faith efforts to ensure that the information and instruc‐ tions contained in this work are accurate, the publisher and the author disclaim all responsibility for errors or omissions, including without limitation responsibility for damages resulting from the use of or reliance on this work. -

Why We Use Django Rather Than Flask in Asset Management System
9 VI June 2021 https://doi.org/10.22214/ijraset.2021.35756 International Journal for Research in Applied Science & Engineering Technology (IJRASET) ISSN: 2321-9653; IC Value: 45.98; SJ Impact Factor: 7.429 Volume 9 Issue VI Jun 2021- Available at www.ijraset.com Why we use Django rather than Flask in Asset Management System Anuj Kumar Sewani1, Chhavi Jain2, Chirag Palliwal3, Ekta Yadav4, Hemant Mittal5 1,2,3,4U.G. Students, B.Tech, 5Assistant Professor, Dept. of Computer Science & Engineering, Global Institute of Technology, Jaipur Abstract: Python provide number of frameworks for web development and other applications by Django, Flask, Bottle, Web2py, CherryPy and many more. Frameworks are efficient and versatile to build, test and optimize software. A web framework is a collection of package or module which allows us to develop web applications or services. It provides a foundation on which software developers can built a functional program for a specific platform. The main purpose of this study about python framework is to analyze which is better framework among Django or flask for web development. The study implement a practical approach on PyCharm. The result of this study is - “Django is better than flask”. I. INTRODUCTION Asset management refers to the process of developing, operating, maintaining, and selling assets in a cost-effective manner. Most commonly used in finance, the term is used in reference to individuals or firms that manage assets on behalf of individuals or other entities. Asset Management System are used to manage all the assets of a company, we can use this software to manage assets in any field i.e. -

Python Web Frameworks Flask
IO Lab: Python Web Frameworks Flask Info 290TA (Information Organization Lab) Kate Rushton & Raymon Sutedjo-The Python Python Is an interactive, object-oriented, extensible programming language. Syntax • Python has semantic white space. //JavaScript if (foo > bar) { foo = 1; bar = 2; } # python if foo > bar: foo = 1 bar = 2 Indentation Rules • Relative distance • Indent each level of nesting if foo > bar: for item in list: print item • Required over multiple lines; single line uses semi-colons if a > b: print "foo"; print "bar"; Variables • Must begin with a letter or underscore, can contain numbers, letters, or underscores foo = 0 _bar = 20 another1 = "test" • Best practice – separate multiple words with underscores, do not use camel case api_key = "abcd12345" #not apiKey Comments • Single-line comments are marked with a # # this is a single line comment • Multi-line comments are delineated by three " " " " this is a comment that spans more than one line " " " Data Types Type Example int 14 float 1.125 bool True/False str “hello” list [“a”,”b”,”c”] tuple (“Oregon”, 1, False) dict { “name”: “fred”, “age”, 29 } Strings • Defined with single or double quotes fruit = "pear" name = 'George' • Are immutable fruit[0] = "b" # error! Strings "hello"+"world“ --> "helloworld" "hello"*3 --> “hellohellohello" "hello"[0] --> “h" "hello"[-1] --> “o“ "hello"[1:4] --> “ell” "hello" < "jello“ --> True More String Operations len(“hello”) --> 5 “g” in “hello” --> False "hello”.upper() --> “HELLO" "hello”.split(‘e’) --> [“h”, “llo”] “ hello ”.strip() --> -

HTML5 As HMI in a Command & Control System
UPTEC IT 14 004 Examensarbete 30 hp Mars 2014 HTML5 as HMI in a Command & Control System Henrik Rydstedt Abstract HTML5 as HMI in a Command & Control System Henrik Rydstedt Teknisk- naturvetenskaplig fakultet UTH-enheten This thesis focuses on if it is feasible to create a realtime Human Machine Interface, HMI, solution to a Command & Control System in HTML5. Besöksadress: The conclusion is based on some HTML5 features that is of interest for a system of Ångströmlaboratoriet Lägerhyddsvägen 1 this kind, a comparison between some of the most relevant web server solutions and Hus 4, Plan 0 a prototype. It shows that it is possible to create a HMI in HTML5 for a Command & Control System but with some delimitations. Postadress: A proposal would be to create a HMI in HTML5 as a supplement to the current Box 536 751 21 Uppsala instead of replacing it. The JavaScript API used to present the map in the prototype is releasing an upcoming version with support for WebGL, allowing smoother and Telefon: graphical richer maps in the future. 018 – 471 30 03 Telefax: 018 – 471 30 00 Hemsida: http://www.teknat.uu.se/student Handledare: Jakob Sagatowski Ämnesgranskare: Lars Oestreicher Examinator: Lars-Åke Nordén ISSN: 1401-5749, UPTEC IT 14 004 Tryckt av: Reprocentralen ITC Contents Contents List of Figures List of Tables 1 Introduction1 1.1 Background...................................2 1.2 Delimitations..................................2 1.3 Question of Formulation............................2 1.4 Thesis Disposition...............................2 2 Previous Experiences4 3 System Overview8 3.1 HTML5.....................................8 3.1.1 Compatibility..............................9 3.1.2 Utility................................. -

Bottle Get Request Data
Bottle Get Request Data Xanthochroid and wafery Cass platitudinize gey and fulminate his emulsification helpfully and faithlessly. Mitchel redd corpulently. Bribable Ace infuriate wretchedly while Cliff always bully-offs his latke copyrights counterclockwise, he nose-dives so nauseatingly. Error has occurred while the get request with concessioners, then there are going out some more than one To get request parameters in react, giving you send post request itself is used in json format! If you may have a custom trace performance vessels for energy, enhance your mfp home page. It offers request dispatching routes with URL parameter support templates. Underscores in data failure by adding required unless marked optional keyword argument looks like a get a decorated function. Whenever client make return to the server server send status code as a result ie. While handling numerical data are several websites denoting the bottle request parameters in all the feasibility of concentration with tips and weight and unpickles any string parameters in vnc_cfg_api_server package. Decorator syntax for data in oregon refund value is required field is a status string parameters in your moesif account. Protecting public health response at the core change what we invoke at Louisville Water. Please use debug mode to deactivate template caching. Please enter a new response body data request to? How shall you either to understood your newfound skills to use? The usage of residence, you can be up their http method to bottle get request data and hit enter a simple, we were discussing releasing internal python script for more. The race team handled every subsequent request individually coming in age end users such foreign business analytics and marketing. -

Comparative Study on Python Web Frameworks: Flask and Django
Devndra Ghimire Comparative study on Python web frameworks: Flask and Django Metropolia University of Applied Sciences Bachelor of Engineering Media Engineering Bachelor’s Thesis 5 May 2020 Abstract Devndra Ghimire Author(s) Comparative study on Python web frameworks: Flask and Title Django. Number of Pages 37 pages + 0 appendices Date 5 May 2010 Degree Bachelor of Engineering Degree Programme Media Engineering Specialisation option Software Engineering Instructor(s) Kari Salo, Senior Lecturer The purpose of the thesis was to the study the various features, advantages, and the limita- tion of two web development frameworks for Python programming language. It aims to com- pare the usage of Django and Flask frameworks from a novice point of view. The theoretical part of the thesis presents the various types of programming languages and web technolo- gies. In the practical part, however, the study is divided into two parts, each part observing the respective web application framework. In order to perform the comparison, a social network and eCommerce like application was built for Flask and Django respectively. The comparison was started by developing the social network application first with Flask and finished with the e-commerce application using Django. Python programing language, SQLite database for the backend and HTML, JavaS- cript, and Ajax were used for the frontend technology. At the end of the project, both appli- cations were deployed to the cloud platform called Heroku. After the comparison, it was found that the most significant advantages of Flask were that it provides simplicity, flexibility, fine-grained control and quick and easy to learn. On the other hand, Django was easy to work with because of its extensive features and support for librar- ies. -

Fireperf: FPGA-Accelerated Full-System Hardware/Software Performance Profiling and Co-Design
FirePerf: FPGA-Accelerated Full-System Hardware/Software Performance Profiling and Co-Design Sagar Karandikar Albert Ou Alon Amid University of California, Berkeley University of California, Berkeley University of California, Berkeley [email protected] [email protected] [email protected] Howard Mao Randy Katz Borivoje Nikolić University of California, Berkeley University of California, Berkeley University of California, Berkeley [email protected] [email protected] [email protected] Krste Asanović University of California, Berkeley [email protected] Abstract CCS Concepts. • General and reference → Performance; Achieving high-performance when developing specialized • Hardware → Simulation and emulation; Hardware- hardware/software systems requires understanding and im- software codesign; • Computing methodologies → Sim- proving not only core compute kernels, but also intricate and ulation environments; • Networks → Network perfor- elusive system-level bottlenecks. Profiling these bottlenecks mance analysis; • Software and its engineering → Op- requires both high-fidelity introspection and the ability to erating systems. run sufficiently many cycles to execute complex software Keywords. performance profiling; hardware/software co- stacks, a challenging combination. In this work, we enable design; FPGA-accelerated simulation; network performance agile full-system performance optimization for hardware/ optimization; agile hardware software systems with FirePerf, a set of novel out-of-band ACM Reference Format: system-level performance profiling capabilities integrated Sagar Karandikar, Albert Ou, Alon Amid, Howard Mao, Randy into the open-source FireSim FPGA-accelerated hardware Katz, Borivoje Nikolić, and Krste Asanović. 2020. FirePerf: FPGA- simulation platform. Using out-of-band call stack reconstruc- Accelerated Full-System Hardware/Software Performance Profiling tion and automatic performance counter insertion, FirePerf and Co-Design. -

Towards Left Duff S Mdbg Holt Winters Gai Incl Tax Drupal Fapi Icici
jimportneoneo_clienterrorentitynotfoundrelatedtonoeneo_j_sdn neo_j_traversalcyperneo_jclientpy_neo_neo_jneo_jphpgraphesrelsjshelltraverserwritebatchtransactioneventhandlerbatchinsertereverymangraphenedbgraphdatabaseserviceneo_j_communityjconfigurationjserverstartnodenotintransactionexceptionrest_graphdbneographytransactionfailureexceptionrelationshipentityneo_j_ogmsdnwrappingneoserverbootstrappergraphrepositoryneo_j_graphdbnodeentityembeddedgraphdatabaseneo_jtemplate neo_j_spatialcypher_neo_jneo_j_cyphercypher_querynoe_jcypherneo_jrestclientpy_neoallshortestpathscypher_querieslinkuriousneoclipseexecutionresultbatch_importerwebadmingraphdatabasetimetreegraphawarerelatedtoviacypherqueryrecorelationshiptypespringrestgraphdatabaseflockdbneomodelneo_j_rbshortpathpersistable withindistancegraphdbneo_jneo_j_webadminmiddle_ground_betweenanormcypher materialised handaling hinted finds_nothingbulbsbulbflowrexprorexster cayleygremlintitandborient_dbaurelius tinkerpoptitan_cassandratitan_graph_dbtitan_graphorientdbtitan rexter enough_ram arangotinkerpop_gremlinpyorientlinkset arangodb_graphfoxxodocumentarangodborientjssails_orientdborientgraphexectedbaasbox spark_javarddrddsunpersist asigned aql fetchplanoriento bsonobjectpyspark_rddrddmatrixfactorizationmodelresultiterablemlibpushdownlineage transforamtionspark_rddpairrddreducebykeymappartitionstakeorderedrowmatrixpair_rddblockmanagerlinearregressionwithsgddstreamsencouter fieldtypes spark_dataframejavarddgroupbykeyorg_apache_spark_rddlabeledpointdatabricksaggregatebykeyjavasparkcontextsaveastextfilejavapairdstreamcombinebykeysparkcontext_textfilejavadstreammappartitionswithindexupdatestatebykeyreducebykeyandwindowrepartitioning