Studies Towards the Total Synthesis of Eleutherobin and Other Marine Natural Products
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Methanesulfonyl Chloride
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -

The Development and Application of Metal-Catalyzed Processes for Organic Synthesis
The Development and Application of Metal-Catalyzed Processes for Organic Synthesis by Edward J. Hennessy B.A. Chemistry Northwestern University, 2000 Submitted to the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY IN ORGANIC CHEMISTRY at the Massachusetts Institute of Technology June 2005 © 2005 Massachusetts Institute of Technology All Rights Reserved Signature of Author: " - - 1_"' .Da-uirnt of Chemistry May 19, 2005 Certified by: Stephen L. Buchwald Camille Dreyfus Professor of Chemistry Thesis Supervisor Accepted by: Robert W. Field Chairman, Departmental Committee on Graduate Students ARCHIVES: This doctoral thesis has been examined by a committee of the Department of Chemistry as follows: -/ Professor Gregory C. Fu: " Ch Chair Professor Stephen L. Buchwald Thesis Supervisor Professor Timothy M. Swager: , -L i \ O 2 The Development and Application of Metal-Catalyzed Processes for Organic Synthesis by Edward J. Hennessy Submitted to the Department of Chemistry on May 19, 2005 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy at the Massachusetts Institute of Technology ABSTRACT Chapter 1. Copper-Catalyzed Arylation of Stabilized Carbanions A mild, general catalytic system for the synthesis of a-aryl malonates has been developed. Aryl iodides bearing a variety of functional groups can be effectively coupled to diethyl malonate in high yields using inexpensive and widely available reagents, making this a superior method to those previously described that employ copper reagents or catalysts. The functional group tolerance of the process developed makes it complementary to analogous palladium-catalyzed couplings. Importantly, a set of mild reaction conditions has been developed that minimize product decomposition, a problem that had not been addressed previously in the literature. -

Nucleophilic Aromatic Substitution
NUCLEOPHILIC AROMATIC SUBSTITUTION Ms. Prerana Sanas M.Pharm (Pharmceutical Chemistry) Asst.Professor, NCRD’s Sterling Institute of Pharmacy Nerul Navi Mumbai Nucleophilic aromatic substitution results in the substitution of a halogen X on a benzene ring by a nucleophile (:Nu– ). Aryl halides undergo a limited number of substitution reactions with strong nucleophiles. NAS occurs by two mechanisms i) Bimoleccular displacement (Addition –Elimination) ii) Benzyne Formation( Elimination –Addition) 7/5/2019 Ms.Prerana Sanas 2 Bimolecular displacement (Addition – Elimination) Aryl halides with strong electron-withdrawing groups (such as NO2) on the ortho or para positions react with nucleophiles to afford substitution products. For example, treatment of p-chloronitrobenzene with hydroxide (– OH) affords p-nitrophenol by replacement of Cl by OH. Nucleophilic aromatic substitution occurs with a variety of strong nucleophiles, including – OH, – OR, – NH2, – SR, and in some cases, neutral nucleophiles such as NH3 and RNH2 . 7/5/2019 Ms.Prerana Sanas 3 Mechanism…… The mechanism of these reactions has two steps: Step i) Addition of the nucleophile (:Nu– ) forms a resonance-stabilized carbanion with a new C – Nu bond—three resonance structures can be drawn. • Step [1] is rate-determining since the aromaticity of the benzene ring is lost. In Step ii) loss of the leaving group re-forms the aromatic ring. This step is fast because the aromaticity of the benzene ring is restored. 7/5/2019 Ms.Prerana Sanas 4 Factors affecting Bimolecular displacement Increasing the number of electron-withdrawing groups increases the reactivity of the aryl halide. Electron-withdrawing groups stabilize the intermediate carbanion, and by the Hammond postulate, lower the energy of the transition state that forms it. -

United States Patent 0
1 3,667,923 United States Patent 0 ice Patented June 6, 1972 1 2 ‘I have discovered that anhydrous hydrogen cyanide $667,923 reacts readily with a quaternary ammonium borohydride PREPARATION OF LITHIUM, SODIUM AND and with the borohydrides of lithium and sodium at QUATERNARY AMMONIUM CYANOBORO atmospheric pressure in solvents for the borohydride, such HYDRIDES Robert C. Wade, Ipswich, Mass, assignor to Ventron as glyme, diglyme, triglyme, tetrahydrofuran and dimethyl Corporation, Beverly, Mass. formamide, or mixtures of these at a temperatures be No Drawing. Filed June 16, 1969, Ser. No. 833,722 tween 0° and the boiling point of the solvent to form Int. Cl. C01c 3/08; C01!) 35/00 the corresponding cyanoborohydride. The preferred sol US. Cl. 23-358 4 Claims vents are tetrahydrofuran and glyme because of their 10 convenient boiling points. Potassium borohydride does not react readily with ABSTRACT OF THE DISCLOSURE hydrogen cyanide in tetrahydrofuran or glyme presum The invention relates to the preparation of lithium, ably because of its lack of solubility in these solvents. sodium and quaternary ammonium cyanoborohydrides. However, it reacts with hydrogen cyanide in dimethyl These compounds are prepared by mixing substantially formamide similarly to the other borohydrides mentioned anhydrous hydrogen cyanide with a substantially an above. hydrous lithium or sodium or quaternary ammonium The reaction of the process of the invention appears to borohydride at a temperature between 0° C. and 100° C. take place in two stages. Thus, if the reaction mixture is in a substantially anhydrous solvent, such as tetrahydro initially maintained between about 10° and about 35° C. -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

Part I. the Total Synthesis Of
AN ABSTRACT OF THE THESIS OF Lester Percy Joseph Burton forthe degree of Doctor of Philosophy in Chemistry presentedon March 20, 1981. Title: Part 1 - The Total Synthesis of (±)-Cinnamodialand Related Drimane Sesquiterpenes Part 2 - Photochemical Activation ofthe Carboxyl Group Via NAcy1-2-thionothiazolidines Abstract approved: Redacted for privacy DT. James D. White Part I A total synthesis of the insect antifeedant(±)-cinnamodial ( ) and of the related drimanesesquiterpenes (±)-isodrimenin (67) and (±)-fragrolide (72)are described from the diene diester 49. Hydro- boration of 49 provided the C-6oxygenation and the trans ring junction in the form of alcohol 61. To confirm the stereoselectivity of the hydroboration, 61 was convertedto both (t)-isodrimenin (67) and (±)-fragrolide (72) in 3 steps. A diisobutylaluminum hydride reduction of 61 followed by a pyridiniumchlorochromate oxidation and treatment with lead tetraacetate yielded the dihydrodiacetoxyfuran102. The base induced elimination of acetic acid preceded theepoxidation and provided 106 which contains the desired hydroxy dialdehydefunctionality of cinnamodial in a protected form. The epoxide 106 was opened with methanol to yield the acetal 112. Reduction, hydrolysis and acetylation of 112 yielded (t)- cinnamodial in 19% overall yield. Part II - Various N- acyl- 2- thionothiazolidineswere prepared and photo- lysed in the presence of ethanol to provide the corresponding ethyl esters. The photochemical activation of the carboxyl function via these derivatives appears, for practical purposes, to be restricted tocases where a-keto hydrogen abstraction and subsequent ketene formation is favored by acyl substitution. Part 1 The Total Synthesis of (±)-Cinnamodial and Related Drimane Sesquiterpenes. Part 2 Photochemical Activation of the Carboxyl Group via N-Acy1-2-thionothiazolidines. -

The Total Synthesis of Securinine and Other Methodology Studies
University of Windsor Scholarship at UWindsor Electronic Theses and Dissertations Theses, Dissertations, and Major Papers 2010 The total synthesis of securinine and other methodology studies Bhartesh Dhudshia University of Windsor Follow this and additional works at: https://scholar.uwindsor.ca/etd Recommended Citation Dhudshia, Bhartesh, "The total synthesis of securinine and other methodology studies" (2010). Electronic Theses and Dissertations. 8275. https://scholar.uwindsor.ca/etd/8275 This online database contains the full-text of PhD dissertations and Masters’ theses of University of Windsor students from 1954 forward. These documents are made available for personal study and research purposes only, in accordance with the Canadian Copyright Act and the Creative Commons license—CC BY-NC-ND (Attribution, Non-Commercial, No Derivative Works). Under this license, works must always be attributed to the copyright holder (original author), cannot be used for any commercial purposes, and may not be altered. Any other use would require the permission of the copyright holder. Students may inquire about withdrawing their dissertation and/or thesis from this database. For additional inquiries, please contact the repository administrator via email ([email protected]) or by telephone at 519-253-3000ext. 3208. The Total Synthesis of Securinine and Other Methodology Studies by Bhartesh Dhudshia A Dissertation Submitted to the Faculty of Graduate Studies through the Department of Chemistry and Biochemistry in Partial Fulfillment of the Requirements -

Organic Synthesis: New Vistas in the Brazilian Landscape
Anais da Academia Brasileira de Ciências (2018) 90(1 Suppl. 1): 895-941 (Annals of the Brazilian Academy of Sciences) Printed version ISSN 0001-3765 / Online version ISSN 1678-2690 http://dx.doi.org/10.1590/0001-3765201820170564 www.scielo.br/aabc | www.fb.com/aabcjournal Organic Synthesis: New Vistas in the Brazilian Landscape RONALDO A. PILLI and FRANCISCO F. DE ASSIS Instituto de Química, UNICAMP, Rua José de Castro, s/n, 13083-970 Campinas, SP, Brazil Manuscript received on September 11, 2017; accepted for publication on December 29, 2017 ABSTRACT In this overview, we present our analysis of the future of organic synthesis in Brazil, a highly innovative and strategic area of research which underpins our social and economical progress. Several different topics (automation, catalysis, green chemistry, scalability, methodological studies and total syntheses) were considered to hold promise for the future advance of chemical sciences in Brazil. In order to put it in perspective, contributions from Brazilian laboratories were selected by the citations received and importance for the field and were benchmarked against some of the most important results disclosed by authors worldwide. The picture that emerged reveals a thriving area of research, with new generations of well-trained and productive chemists engaged particularly in the areas of green chemistry and catalysis. In order to fulfill the promise of delivering more efficient and sustainable processes, an integration of the academic and industrial research agendas is to be expected. On the other hand, academic research in automation of chemical processes, a well established topic of investigation in industrial settings, has just recently began in Brazil and more academic laboratories are lining up to contribute. -

덴드리틱 벤질 클로라이드의 효율적인 합성 Facile Synthesis of Dendritic Benzyl Chlorides from Their Alcohols W
Polymer(Korea), Vol. 31, No. 5, pp 417-421, 2007 덴드리틱 벤질 클로라이드의 효율적인 합성 이재욱F,7ㆍ한승철Fㆍ김희주ㆍ김정환ㆍ이언엽ㆍ김병기ㆍ성새름ㆍ강화신ㆍ김지현FFㆍ허도성FFF 동아대학교 화학과, F동아대학교 의생명과학과, FF동국대학교 생명화학공학과, FFF인제대학교 의생명화학과 (2007년 5월 9일 접수, 2007년 6월 29일 채택) Facile Synthesis of Dendritic Benzyl Chlorides from Their Alcohols with Methanesulfonyl Chloride/Et3N Jae Wook Lee*,7, Seung Choul Han*, Hee Joo Kim, Jung Hwan Kim, Un Yup Lee, Byoung-Ki Kim, Sae Reum Sung, Hwa-Shin Kang, Ji Hyeon Kim**, and Do-Sung Huh*** Department of Chemistry, Dong-A University, Hadan-2-dong, Busan 604-714, Korea *Department of Medical Bioscience, Dong-A University, Hadan-2-dong, Busan 604-714, Korea **Department of Chemical & Biochemical Engineering, Dongguk University, Seoul 100-715, Korea ***Department of Biomedicinal Chemistry, Inje University, Gim Hai 621-749, Korea (Received May 9, 2007; Accepted June 29, 2007) 초록: 덴드리틱 벤질 알코올을 트리에틸아민과 메탄술포닐클로라이드와 반응시켜서, 덴드리틱 벤질 클로라이드 의 효율적인 합성이 이루어졌다. 이 반응은 히드록시기의 메실화 반응과 염소화 반응의 2단계 반응으로 이루어지 는데, 중간체의 분리없이 한 반응 용기내에서 반응이 진행되는 경제적인 방법이다. Abstract: A successful rapid synthesis of dendritic benzyl chlorides from dendritic benzyl alcohols using methanesulfonyl chloride/Et3N as activating agents was described. In this method, each dendritic benzyl chloride can be prepared in one pot: no isolation of intermediate mesylated dendrons is required. The key steps in the syntheses of dendritic benzyl chlorides were the mesylation of the hydroxymethyl group followed by the chlorination by in-situ generated triethylammonium chloride. Keywords: benzylic alcohol, chlorination. Introduction vation and coupling steps in high yield. In addition, the coupling step must be very efficient to enable complete Dendrimers represent a novel type of polymeric material reaction. -
![Reductive Amination with [11C]Formaldehyde: a Versatile Approach to Radiomethylation of Amines](https://docslib.b-cdn.net/cover/4140/reductive-amination-with-11c-formaldehyde-a-versatile-approach-to-radiomethylation-of-amines-684140.webp)
Reductive Amination with [11C]Formaldehyde: a Versatile Approach to Radiomethylation of Amines
International Journal of Organic Chemistry, 2012, 2, 202-223 http://dx.doi.org/10.4236/ijoc.2012.23030 Published Online September 2012 (http://www.SciRP.org/journal/ijoc) Reductive Amination with [11C]Formaldehyde: A Versatile Approach to Radiomethylation of Amines Chunying Wu1, Ruoshi Li1, Dorr Dearborn2, Yanming Wang1* 1Division of Radiopharmaceutical Science, Case Center for Imaging Research, Department of Radiology, Cleveland, USA 2Environmental Health, Case Western Reserve University, Cleveland, USA Email: *[email protected] Received February 16, 2012; revised March 24, 2012; accepted April 2, 2012 ABSTRACT Carbon-11 radiolabeled amines constitute a very important class of radioligands that are widely used for positron emis- 11 sion tomography (PET) imaging. Radiolabeling of amines is often achieved through radiomethylation using [ C]CH3I 11 or [ C]CH3OTf under basic conditions in a strictly anhydrous environment. Functional groups such as hydroxyl and carboxyl groups that are often present in the molecules are normally base sensitive and require protection and deprotec- tion, which substantially prolongs and complicates the radiolabeling process. Here we report a versatile approach to a series of C-11 radiolabeled amines prepared through reductive amination using [11C]formaldehyde. Using a variety of substrates bearing different functional groups, we demonstrate the general utility of this method. In contrast to conven- tional radiomethylation methods, the reductive amination using [11C]formaldehyde can be carried out in an aqueous environment relatively quickly without the need of protection of base-sensitive functional groups. Keywords: C-11 Formaldehyde; Radiomethylation; Reductive Amination; Positron Emission Tomography; Radiolabelling 1. Introduction probes must be labeled with positron-emitting radionu- clides such as carbon-11 or fluorine-18. -
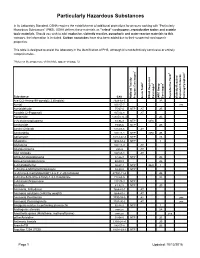
Particularly Hazardous Substances
Particularly Hazardous Substances In its Laboratory Standard, OSHA requires the establishment of additional protections for persons working with "Particularly Hazardous Substances" (PHS). OSHA defines these materials as "select" carcinogens, reproductive toxins and acutely toxic materials. Should you wish to add: explosive, violently reactive, pyrophoric and water-reactve materials to this category, the information is included. Carbon nanotubes have also been added due to their suspected carcinogenic properties. This table is designed to assist the laboratory in the identification of PHS, although it is not definitively conclusive or entirely comprehensive. *Notes on the proper use of this table appear on page 12. 1 6 5 2 3 4 Substance CAS National Toxicity National Program Carcinogen Toxin Acute Regulated OSHA Carcinogen Group IARC Carcinogen Toxin Reproductive Violently Reactive/ Explosive/Peroxide Forming/Pyrophoric A-a-C(2-Amino-9H-pyrido[2,3,b]indole) 2648-68-5 2B Acetal 105-57-7 yes Acetaldehyde 75-07-0 NTP AT 2B Acrolein (2-Propenal) 107-02-8 AT Acetamide 126850-14-4 2B 2-Acetylaminofluorene 53-96-3 NTP ORC Acrylamide 79-06-6 NTP 2B Acrylyl Chloride 814-68-6 AT Acrylonitrile 107-13-1 NTP ORC 2B Adriamycin 23214-92-8 NTP 2A Aflatoxins 1402-68-2 NTP 1 Allylamine 107-11-9 AT Alkylaluminums varies AT Allyl Chloride 107-05-1 AT ortho-Aminoazotoluene 97-56-3 NTP 2B para-aminoazobenzene 60-09-3 2B 4-Aminobiphenyl 92-67-1 NTP ORC 1 1-Amino-2-Methylanthraquinone 82-28-0 NTP (2-Amino-6-methyldipyrido[1,2-a:3’,2’-d]imidazole) 67730-11-4 2B -

Approaches Towards the Synthesis of Bradyoxetin
University of Mississippi eGrove Electronic Theses and Dissertations Graduate School 2010 Approaches Towards the Synthesis of Bradyoxetin Vanildo Martins Lima Braga Follow this and additional works at: https://egrove.olemiss.edu/etd Part of the Organic Chemistry Commons Recommended Citation Braga, Vanildo Martins Lima, "Approaches Towards the Synthesis of Bradyoxetin" (2010). Electronic Theses and Dissertations. 61. https://egrove.olemiss.edu/etd/61 This Dissertation is brought to you for free and open access by the Graduate School at eGrove. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of eGrove. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Vanildo Martins L. Braga entitled “Approaches Towards the Synthesis of Bradyoxetin. ” I have examined the final copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Pharmaceutical Sciences , with an emphasis in Medicinal Chemistry . ______________________________ John M. Rimoldi, Major Professor Professor of Medical Chemistry We have read this dissertation and recommend its acceptance: _____________________________________ Mitchell A. Avery Professor of Medicinal Chemistry _____________________________________ Franck E. Dayan Adjunct Professor of Medicinal Chemistry _____________________________________ Marc Slattery Professor of Pharmacognosy Accepted for the Council: ____________________________ Dean of the Graduate School STATEMENT OF PERMISSION TO USE In presenting this thesis in partial fulfillment of the requirements for a Ph.D. degree at The University of Mississippi, I agree that the Library shall make it available to borrowers under rules of the Library.