13Balkan.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DIAGMOL Town, ZIPCODE :…………………………………………………… Director : Prof
Mr. Mrs. (IN UPPER CASE, please) Name:……………………………………………………. Maiden Name:……………………………………. DMGL / Service de Médecine Génétique Centre d’accueil des prélèvements (CAP) First name :………………………………………………… Bâtiment des Laboratoires (BATLab), local 8D-0-850.1 Date of birth : ............ / ........... / …………… 4 rue Gabrielle-Perret-Gentil, 1211 Genève 14 Legal representative (for minors) : father mother Molecular and Genomic Diagnostics Laboratory Name/first name :…………………………………………… Street/N°:……………………………………………………… http://www.hug-ge.ch/feuilles-de-demande DIAGMOL Town, ZIPCODE :…………………………………………………… Director : Prof. Stylianos ANTONARAKIS Hospitalisation Unit: …………… Physician :…………………… Lab managers: N° EdS : …………………………………………………………… Dr J.-L. BLOUIN, Dr Th. NOUSPIKEL, Dr. M. GUIPPONI Invoice address: Patient Prescriptor Insurance [email protected], [email protected], [email protected] Type of case : Disease AI Accident Pregnancy Lab direct or results: Phone/FAX: +41 (0) 22 37 21 826 / 21 860 N° AVS (Mandatory for AI) : ………………………...................... Sample Entrance Center (CAP) : Phone +41 (0) 22 37 21 800 Insurance : ………………… Insured N° : ……………………… PHYSICIAN PHYSICIAN (NAME/First name - Street/N°- Town, ZIPCODE - Phone/FAX. IN UPPER CASES, PLEASE) COPY TO OTHER PHYSICIAN (NAME/First name - Street/N°- Town, ZIPCODE - Phone/FAX. IN UPPER CASES, PLEASE) « The laboratory is granted permission by the Physician/Patient to transmit copies of the report to other physicians» Opposition of the patient to the registration of this request results in the electronic patient record (DPI) of the HUG If the patient belongs to a family already known to the laboratory, please indicate index case NAME: CLINICAL INFORMATIONS given by the physician: Ethnic origins Father Mother Currently pregnant Date of last menses Number of weeks of amenorrhea SAMPLE(S) Most of our tests work from 4 ml of blood in EDTA (children <2 ans : 1 On every and single NAME First name ml : ok) or from purified DNA (some exceptions apply for some tests) tube. -

Neonatal Dermatology Review
NEONATAL Advanced Desert DERMATOLOGY Dermatology Jennifer Peterson Kevin Svancara Jonathan Bellew DISCLOSURES No relevant financial relationships to disclose Off-label use of acitretin in ichthyoses will be discussed PHYSIOLOGIC Vernix caseosa . Creamy biofilm . Present at birth . Opsonizing, antibacterial, antifungal, antiparasitic activity Cutis marmorata . Reticular, blanchable vascular mottling on extremities > trunk/face . Response to cold . Disappears on re-warming . Associations (if persistent) . Down syndrome . Trisomy 18 . Cornelia de Lange syndrome PHYSIOLOGIC Milia . Hard palate – Bohn’s nodules . Oral mucosa – Epstein pearls . Associations . Bazex-Dupre-Christol syndrome (XLD) . BCCs, follicular atrophoderma, hypohidrosis, hypotrichosis . Rombo syndrome . BCCs, vermiculate atrophoderma, trichoepitheliomas . Oro-facial-digital syndrome (type 1, XLD) . Basal cell nevus (Gorlin) syndrome . Brooke-Spiegler syndrome . Pachyonychia congenita type II (Jackson-Lawler) . Atrichia with papular lesions . Down syndrome . Secondary . Porphyria cutanea tarda . Epidermolysis bullosa TRANSIENT, NON-INFECTIOUS Transient neonatal pustular melanosis . Birth . Pustules hyperpigmented macules with collarette of scale . Resolve within 4 weeks . Neutrophils Erythema toxicum neonatorum . Full term . 24-48 hours . Erythematous macules, papules, pustules, wheals . Eosinophils Neonatal acne (neonatal cephalic pustulosis) . First 30 days . Malassezia globosa & sympoidalis overgrowth TRANSIENT, NON-INFECTIOUS Miliaria . First weeks . Eccrine -

Should 45,X/46,XY Boys with No Or Mild Anomaly of External Genitalia
3 179 L Dumeige and others 45,X/46,XY phenotypic boys, 179:3 181–190 Clinical Study not so benign? Should 45,X/46,XY boys with no or mild anomaly of external genitalia be investigated and followed up? Laurence Dumeige1,2, Livie Chatelais3, Claire Bouvattier4, Marc De Kerdanet5, Capucine Hyon6, Blandine Esteva7, Dinane Samara-Boustani8, Delphine Zenaty1, Marc Nicolino9, Sabine Baron10, Chantal Metz-Blond11, Catherine Naud-Saudreau12, Clémentine Dupuis13, Juliane Léger1, Jean-Pierre Siffroi6, Bruno Donadille14, Sophie Christin-Maitre14, Jean-Claude Carel1, Regis Coutant3 and Laetitia Martinerie1,2 1Pediatric Endocrinology Department, CHU Robert Debré, Centre de Référence des Maladies Endocriniennes Rares de la Croissance, Assistance-Publique Hôpitaux de Paris and Université Paris Diderot, Sorbonne Paris Cité, Paris, France, 2INSERM UMR-S1185, Le Kremlin Bicêtre, France, 3Pediatric Department, CHU Angers, Angers, France, 4Pediatric Endocrinology Department, CHU Bicêtre, Centre de Référence des Anomalies du Développement Génital, Assistance-Publique Hôpitaux de Paris, Le Kremlin-Bicêtre, France, 5Pediatric Department, CHU Rennes, Rennes, France, 6Genetic Department, 7Pediatric Endocrinology Department, CHU Armand Trousseau, Centre de Référence des Maladies Endocriniennes Rares de la Croissance, Assistance-Publique Hôpitaux de Paris, Paris, France, 8Pediatric Endocrinology Department, CHU Necker-Enfants Malades, Centre de Référence des Maladies Endocriniennes Rares de la Croissance, Assistance-Publique Hôpitaux de Paris, Paris, France, 9Pediatric -

Genetics of Azoospermia
International Journal of Molecular Sciences Review Genetics of Azoospermia Francesca Cioppi , Viktoria Rosta and Csilla Krausz * Department of Biochemical, Experimental and Clinical Sciences “Mario Serio”, University of Florence, 50139 Florence, Italy; francesca.cioppi@unifi.it (F.C.); viktoria.rosta@unifi.it (V.R.) * Correspondence: csilla.krausz@unifi.it Abstract: Azoospermia affects 1% of men, and it can be due to: (i) hypothalamic-pituitary dysfunction, (ii) primary quantitative spermatogenic disturbances, (iii) urogenital duct obstruction. Known genetic factors contribute to all these categories, and genetic testing is part of the routine diagnostic workup of azoospermic men. The diagnostic yield of genetic tests in azoospermia is different in the different etiological categories, with the highest in Congenital Bilateral Absence of Vas Deferens (90%) and the lowest in Non-Obstructive Azoospermia (NOA) due to primary testicular failure (~30%). Whole- Exome Sequencing allowed the discovery of an increasing number of monogenic defects of NOA with a current list of 38 candidate genes. These genes are of potential clinical relevance for future gene panel-based screening. We classified these genes according to the associated-testicular histology underlying the NOA phenotype. The validation and the discovery of novel NOA genes will radically improve patient management. Interestingly, approximately 37% of candidate genes are shared in human male and female gonadal failure, implying that genetic counselling should be extended also to female family members of NOA patients. Keywords: azoospermia; infertility; genetics; exome; NGS; NOA; Klinefelter syndrome; Y chromosome microdeletions; CBAVD; congenital hypogonadotropic hypogonadism Citation: Cioppi, F.; Rosta, V.; Krausz, C. Genetics of Azoospermia. 1. Introduction Int. J. Mol. Sci. -

Ichthyosis Focus Spring 2002
ICHTHYOSIS FOCUS Vol. 21, No. 1 A Quarterly Journal for Friends of F.I.R.S.T. Spring 2002 Unlocking the Secrets of a Rare Ichthyosis abriele Richard, MD, Assistant Professor of A note from Dr. Richard: Our research group GDermatology and Genetics at the Thomas greatly acknowledges the support of F.I.R.S.T., Jefferson University in Philadelphia, received an along with the ASA, NAAF, and the National American Skin Association Categorical Disease Registry for Ichthyosis and Related Disorders, as Grant for Childhood Skin Disease Research. well as the participation of many families in our Specifically, she studied the genetics of Netherton studies. With the help of a F.I.R.S.T. research award Syndrome (NTS), a rare form of severe ichthyosis (awarded in 1999) we were able to perform genetic associated with other abnormalities. linkage studies to localize the Netherton Syndrome NTS is an inherited disorder that presents at gene and to initiate mutation analysis of SPINK5. birth with generalized redness, thickening and The financial support allowed us to amass a critical scaling of the skin, which may persist through life amount of data to successfully apply for NIH fund- or change into scaly, itchy plaques. The hairs may Gabriele Richard, MD ing of our research. As a result, we were able to be sparse, fragile, and short due to multiple swellings along screen more than 31 NTS families to date, identifying 24 dis- the hair shafts, at which the hair breaks easily. Also charac- tinct SPINK5 mutations in 23 families. In 8 families genetic teristic of NTS is a predisposition to allergies, infections, and testing revealed no mutations in SPINK5, suggesting that these sometimes, failure to thrive. -

Integrating Clinical and Genetic Approaches in the Diagnosis of 46,XY Disorders of Sex Development
ID: 18-0472 7 12 Z Kolesinska et al. Diagnostic approach of 46,XY 7:12 1480–1490 DSD RESEARCH Integrating clinical and genetic approaches in the diagnosis of 46,XY disorders of sex development Zofia Kolesinska1, James Acierno Jr2, S Faisal Ahmed3, Cheng Xu2, Karina Kapczuk4, Anna Skorczyk-Werner5, Hanna Mikos1, Aleksandra Rojek1, Andreas Massouras6, Maciej R Krawczynski5, Nelly Pitteloud2 and Marek Niedziela1 1Department of Pediatric Endocrinology and Rheumatology, Poznan University of Medical Sciences, Poznan, Poland 2Endocrinology, Diabetology & Metabolism Service, Lausanne University Hospital, Lausanne, Switzerland 3Developmental Endocrinology Research Group, School of Medicine, Dentistry & Nursing, University of Glasgow, Glasgow, UK 4Division of Gynecology, Department of Perinatology and Gynecology, Poznan University of Medical Sciences, Poznan, Poland 5Department of Medical Genetics, Poznan University of Medical Sciences, Poznan, Poland 6Saphetor, SA, Lausanne, Switzerland Correspondence should be addressed to M Niedziela: [email protected] Abstract 46,XY differences and/or disorders of sex development (DSD) are clinically and Key Words genetically heterogeneous conditions. Although complete androgen insensitivity f array-comparative syndrome has a strong genotype–phenotype correlation, the other types of 46,XY DSD genomic hybridization are less well defined, and thus, the precise diagnosis is challenging. This study focused f differences and/or disorders of sex on comparing the relationship between clinical assessment and genetic findings in development a cohort of well-phenotyped patients with 46,XY DSD. The study was an analysis of f massive parallel/next clinical investigations followed by genetic testing performed on 35 patients presenting generation sequencing to a single center. The clinical assessment included external masculinization score f oligogenicity (EMS), endocrine profiling and radiological evaluation. -

10Th Anniversary of the Human Genome Project
Grand Celebration: 10th Anniversary of the Human Genome Project Volume 3 Edited by John Burn, James R. Lupski, Karen E. Nelson and Pabulo H. Rampelotto Printed Edition of the Special Issue Published in Genes www.mdpi.com/journal/genes John Burn, James R. Lupski, Karen E. Nelson and Pabulo H. Rampelotto (Eds.) Grand Celebration: 10th Anniversary of the Human Genome Project Volume 3 This book is a reprint of the special issue that appeared in the online open access journal Genes (ISSN 2073-4425) in 2014 (available at: http://www.mdpi.com/journal/genes/special_issues/Human_Genome). Guest Editors John Burn University of Newcastle UK James R. Lupski Baylor College of Medicine USA Karen E. Nelson J. Craig Venter Institute (JCVI) USA Pabulo H. Rampelotto Federal University of Rio Grande do Sul Brazil Editorial Office Publisher Assistant Editor MDPI AG Shu-Kun Lin Rongrong Leng Klybeckstrasse 64 Basel, Switzerland 1. Edition 2016 MDPI • Basel • Beijing • Wuhan ISBN 978-3-03842-123-8 complete edition (Hbk) ISBN 978-3-03842-169-6 complete edition (PDF) ISBN 978-3-03842-124-5 Volume 1 (Hbk) ISBN 978-3-03842-170-2 Volume 1 (PDF) ISBN 978-3-03842-125-2 Volume 2 (Hbk) ISBN 978-3-03842-171-9 Volume 2 (PDF) ISBN 978-3-03842-126-9 Volume 3 (Hbk) ISBN 978-3-03842-172-6 Volume 3 (PDF) © 2016 by the authors; licensee MDPI, Basel, Switzerland. All articles in this volume are Open Access distributed under the Creative Commons License (CC-BY), which allows users to download, copy and build upon published articles even for commercial purposes, as long as the author and publisher are properly credited, which ensures maximum dissemination and a wider impact of our publications. -

UNSCEAR 2001 Report to the General Assembly, with Scientific Annex
HEREDITARY EFFECTS OF RADIATION United Nations Scientific Committee on the Effects of Atomic Radiation UNSCEAR 2001 Report to the General Assembly, with Scientific Annex UNITED NATIONS HEREDITARY EFFECTS OF RADIATION United Nations Scientific Committee on the Effects of Atomic Radiation UNSCEAR 2001 Report to the General Assembly, with Scientific Annex UNITED NATIONS New York, 2001 NOTE The report of the Committee without its scientific annex appears as Official Records of the General Assembly. Fifty-sixth Session, Supplement No. 46 (N56146). The designation employed and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the pad of the Secretariat of the United Nationsconcerning the legal status of any country, temtory, city orarea, or of its authorities, or concerning the delimitation of its frontiers or boundaries. The country names used in this document are, In most cases, those that were in use at the time the data were collected or the text prepared. In othercases, however, the names have been updated, where this was posslble and appropriate, to reflect political changes. UNITED NATIONS PUBLICATION Sales No. E.O1 .IX.2 ISBN 92-1-1 42244-2 Report of the United Nations Scientific Committee on the Effects of Atomic Radiation to the General Assembly 1. During the past few years, the United Nations 4. The Committee wishes to acknowledge the assistance Scientific Committee on the Effects of Atomic Radiation1 of the consultant, K. Sankaranarayanan, in the preparation has undertaken broad reviews of the sources and effects of of the scientific annex and the advice of the international ionizing radiation. -

Uncommon Forms of Diabetes
Clinical Medicine 2021 Vol 21, No 4: e337–41 CME: DIABETES Uncommon forms of diabetes Authors: Yun-Ni LeeA and Mohammed SB HudaB Diabetes mellitus is a common condition which all clinicians and insulin independence. It is estimated to account for 1%–2% will encounter in their clinical practice. The most common of patients diagnosed with diabetes and, in the UK, the prevalence form is type 2 diabetes followed by type 1 diabetes. However, of MODY is estimated to be at 108 cases per million.3 However, there are many other atypical forms of diabetes which are it may be a significant underestimate and these figures are not important for a clinician to consider as it can impact on the accurate until large population screening studies are performed. ABSTRACT diagnosis and their management. The most common mutations are hepatocyte nuclear factor-1- This article focuses on maturity onset diabetes of the young alpha (HNF1α; 52%), glucokinase (GCK; 32%) and HNF4α (10%), (MODY), latent autoimmune diabetes in adults (LADA), see Table 2.3 ketosis-prone diabetes and other secondary forms of diabetes such as pancreatic cancer and haemochromatosis. We briefly Hepatocyte nuclear factor-1-alpha gene describe the key clinical features of these forms of diabetes and their investigations and treatment. Formerly called MODY3, mutations on the HNF1α gene on chromosome 3 are associated with a progressive defect of insulin secretion.4 Mutations here also result in low renal threshold for 5 Introduction glucose and thus mutation carriers have detectable glycosuria. In the UK, around 90% of people with diabetes have type 2 diabetes (T2D), around 8% have type 1 diabetes (T1D) and around 2% have other forms of diabetes.1 Key points Typically, we see T1D present in a young, lean patient with Suspect other uncommon forms of diabetes if the clinical marked symptoms of polyuria, polydipsia, weight loss and diabetic picture does not fit type 1 or type 2 diabetes. -

Mat Kadi Tora Tutti O Al Ut Hit Hitta Atuh
MAT KADI TORA TUTTI USO AL20180235194A1 UT HIT HITTA ATUH ( 19) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0235194 A1 Fahrenkrug et al. (43 ) Pub . Date : Aug . 23, 2018 ( 54 ) MULTIPLEX GENE EDITING Publication Classification (51 ) Int. Ci. ( 71 ) Applicant: Recombinetics , Inc ., Saint Paul, MN A01K 67/ 027 (2006 . 01 ) (US ) C12N 15 / 90 ( 2006 .01 ) (72 ) Inventors : Scott C . Fahrenkrug, Minneapolis , (52 ) U . S . CI. MN (US ) ; Daniel F . Carlson , CPC .. .. A01K 67 / 0276 (2013 . 01 ) ; C12N 15 / 907 Woodbury , MN (US ) ( 2013 .01 ) ; A01K 67 /0275 ( 2013 .01 ) ; A01K 2267/ 02 (2013 .01 ) ; AOIK 2217 / 15 (2013 .01 ) ; AOIK 2227 / 108 ( 2013 .01 ) ; AOIK 2217 /07 (21 ) Appl. No. : 15 /923 , 951 ( 2013 .01 ) ; A01K 2227/ 101 (2013 .01 ) ; AOIK ( 22 ) Filed : Mar. 16 , 2018 2217 /075 ( 2013 .01 ) (57 ) ABSTRACT Related U . S . Application Data Materials and methods for making multiplex gene edits in (62 ) Division of application No . 14 /698 ,561 , filed on Apr. cells and are presented . Further methods include animals 28 , 2015, now abandoned . and methods of making the same . Methods of making ( 60 ) Provisional application No . 61/ 985, 327, filed on Apr. chimeric animals are presented , as well as chimeric animals . 28 , 2014 . Specification includes a Sequence Listing . Patent Application Publication Aug . 23 , 2018 Sheet 1 of 13 US 2018 / 0235194 A1 GENERATION OF HOMOZYGOUS CATTLE EDITED AT ONE ALLELE USING SINGLE EDITS Edit allele , Raise FO to Mate FO enough times Raise F1s to Mate F1 siblings Clone cell , sexual maturity , to produce enough F1 sexualmaturity , to make Implant, Gestate 2 years generation carrying 2 years homozygous KO , 9 months , birth edited allele to mate 9 months of FO with each other Generation Primary Fibroblasts ? ? ? Time, years FIG . -
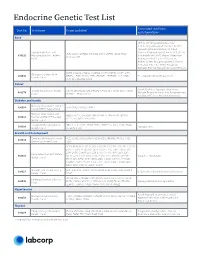
Endocrine Genetic Test List
Endocrine Genetic Test List Associated conditions Test No. Test Name Genes Included and phenotypes Bone HPP, X-Linked Hypophosphatemia, X-Linked Hypophosphatemic Rickets, XLH, Hypophosphatemic Rickets, X-Linked Hypophosphatasia and Dominant Hypophosphatemic Rickets (XLHR), ALPL, CLCN5, CYP2R1, CYP27B1, DMP1, ENPP1, FGF23, PHEX, 630292 Hypophosphatemic Rickets X-Linked Rickets (XLR), Vitamin D-Resistant SLC34A3, VDR Panel Rickets, X-Linked Vitamin D-Resistant Rickets (VDRR), Hypophosphatemic Vitamin D-Resistant Rickets (HPDR), Phosphate Diabetes, Familial Hypophosphatemic Rickets BMP1, COL1A1, COL1A2, CREB3L1, CRTAP, FKBP10, IFITM5, LRP5, Osteogenesis Imperfecta 630543 MBTPS2, P3H1, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, SPARC, OI, Juvenile Primary Osteoporosis Genetic Panel TENT5A, TMEM38B, WNT1 Cancer Familial Isolated Hyperparathyroidism, VistaSeq® Endocrine Cancer CDC73, MAX, MEN1, NF1, PRKAR1A, PTEN, RET, SDHB, SDHC, SDHD, 481374 Multiple Endocrine Neoplasia, Paraganglioma, Panel* TMEM127, TP53 and VHL Parathyroid Cancer, Pheochromocytoma Diabetes and Insulin Maturity-Onset Diabetes of the 630568 GCK, HNF1A, HNF1B, HNF4A Young (MODY) 4-gene Panel Maturity-Onset Diabetes of ABCC8, APPL1, BLK, GCK, HNF1A, HNF1B, HNF4A, INS, KCNJ11, 630513 the Young (MODY) Expanded KLF11, NEUROD1, PAX4, PDX1 Genetic Panel Congenital Hyperinsulinism ABCC8, GCK, GLUD1, HADH, HNF1A, HNF4A, KCNJ11, PGM1, PMM2, 630500 Hypoglycemia Genetic Panel SLC16A1, UCP2 Growth and Development Combined Pituitary Hormone GLI1, HESX1, LHX3, LHX4, OTX2, POU1F1, PROKR2, PROP1, -

Discovery in Genetic Skin Disease: the Impact of High Throughput Genetic Technologies
Genes 2014, 5, 615-634; doi:10.3390/genes5030615 OPEN ACCESS genes ISSN 2073-4425 www.mdpi.com/journal/genes Review Discovery in Genetic Skin Disease: The Impact of High Throughput Genetic Technologies Thiviyani Maruthappu †, Claire A. Scott † and David P. Kelsell * Centre for Cutaneous Research, Blizard Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, 4 Newark Street, London E1 2AT, UK; E-Mails: [email protected] (T.M.); [email protected] (C.A.S.) † These authors contributed equally to this work and should be considered joint first authors. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel: +44-20-7882-7167; Fax: +44-20-7882-7172. Received: 4 April 2014; in revised form: 7 July 2014 / Accepted: 14 July 2014 / Published: 4 August 2014 Abstract: The last decade has seen considerable advances in our understanding of the genetic basis of skin disease, as a consequence of high throughput sequencing technologies including next generation sequencing and whole exome sequencing. We have now determined the genes underlying several monogenic diseases, such as harlequin ichthyosis, Olmsted syndrome, and exfoliative ichthyosis, which have provided unique insights into the structure and function of the skin. In addition, through genome wide association studies we now have an understanding of how low penetrance variants contribute to inflammatory skin diseases such as psoriasis vulgaris and atopic dermatitis, and how they contribute to underlying pathophysiological disease processes. In this review we discuss strategies used to unravel the genes underlying both monogenic and complex trait skin diseases in the last 10 years and the implications on mechanistic studies, diagnostics, and therapeutics.