Global Landscape of Phenazine Biosynthesis And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Growth and Adaptation of Microorganisms on the Cheese Surface Christophe Monnet, Sophie Landaud-Liautaud, Pascal Bonnarme, Dominique Swennen
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Archive Ouverte en Sciences de l'Information et de la Communication Growth and adaptation of microorganisms on the cheese surface Christophe Monnet, Sophie Landaud-Liautaud, Pascal Bonnarme, Dominique Swennen To cite this version: Christophe Monnet, Sophie Landaud-Liautaud, Pascal Bonnarme, Dominique Swennen. Growth and adaptation of microorganisms on the cheese surface. FEMS Microbiology Letters, Wiley-Blackwell, 2015, 362 (1), pp.1-9. 10.1093/femsle/fnu025. hal-01535275 HAL Id: hal-01535275 https://hal.archives-ouvertes.fr/hal-01535275 Submitted on 28 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 1 Growth and adaptation of microorganisms on the cheese surface 2 3 4 Christophe Monnet1,2, Sophie Landaud2,1, Pascal Bonnarme1,2 & Dominique Swennen3,4 5 6 1 INRA, UMR782 Génie et Microbiologie des Procédés Alimentaires, 78370 Thiverval- 7 Grignon, France 8 2 AgroParisTech, UMR782 Génie et Microbiologie des Procédés Alimentaires, 78370 9 Thiverval-Grignon, France 10 3 INRA, UMR1319 Micalis, 78370 Thiverval-Grignon, France 11 4 AgroParisTech, UMR1319 Micalis, 78370 Thiverval-Grignon, France 12 13 * Corresponding author. -

Exploring the Diversity and Antimicrobial Potential of Marine Actinobacteria from the Comau Fjord in Northern Patagonia, Chile
ORIGINAL RESEARCH published: 19 July 2016 doi: 10.3389/fmicb.2016.01135 Exploring the Diversity and Antimicrobial Potential of Marine Actinobacteria from the Comau Fjord in Northern Patagonia, Chile Agustina Undabarrena 1, Fabrizio Beltrametti 2, Fernanda P. Claverías 1, Myriam González 1, Edward R. B. Moore 3, 4, Michael Seeger 1 and Beatriz Cámara 1* 1 Laboratorio de Microbiología Molecular y Biotecnología Ambiental, Departamento de Química & Centro de Biotecnología Daniel Alkalay Lowitt, Universidad Técnica Federico Santa María, Valparaíso, Chile, 2 Actygea S.r.l., Gerenzano, Italy, 3 Culture Collection University of Gothenburg (CCUG), Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden, 4 Department of Infectious Diseases, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden Edited by: Bioprospecting natural products in marine bacteria from fjord environments are attractive Learn-Han Lee, due to their unique geographical features. Although, Actinobacteria are well known Monash University Malaysia Campus, Malaysia for producing a myriad of bioactive compounds, investigations regarding fjord-derived Reviewed by: marine Actinobacteria are scarce. In this study, the diversity and biotechnological Atte Von Wright, potential of Actinobacteria isolated from marine sediments within the Comau University of Eastern Finland, Finland fjord, in Northern Chilean Patagonia, were assessed by culture-based approaches. Polpass Arul Jose, Central Salt and Marine Chemicals The 16S rRNA gene sequences revealed that members phylogenetically related Research Institute, India to the Micrococcaceae, Dermabacteraceae, Brevibacteriaceae, Corynebacteriaceae, *Correspondence: Microbacteriaceae, Dietziaceae, Nocardiaceae, and Streptomycetaceae families were Beatriz Cámara [email protected] present at the Comau fjord. A high diversity of cultivable Actinobacteria (10 genera) was retrieved by using only five different isolation media. -

UNIVERSITY of CALIFORNIA RIVERSIDE Gustatory Receptors In
UNIVERSITY OF CALIFORNIA RIVERSIDE Gustatory Receptors in Mosquito Olfaction and Host-Seeking Behavior A Dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Entomology by Genevieve Mitchell Tauxe March 2015 Dissertation Committee: Dr. Anandasankar Ray, Chairperson Dr. Michael E. Adams Dr. Ring T. Cardé Dr. Jocelyn G. Millar Copyright by Genevieve Mitchell Tauxe 2015 The Dissertation of Genevieve Mitchell Tauxe is approved: Committee Chairperson University of California, Riverside Acknowledgements First, I thank the anonymous odor donors who participated in the experiments described here. They variously washed their feet in the bathroom sink, walked around for hours with beads in their socks (which, no, is not terribly comfortable), and continually showed up no matter how many times we kept asking for more socks. Thank you for your generosity, and for your stinky feet. Now go find yourself on page 73. I thank my advisor, Anand Ray, for all of his help and support over my graduate career. He has taught me so much about how to do research, and also how to compose manuscripts, give presentations, and talk about science. I also thank my committee members, Ring Cardé, Jocelyn Millar, and Mike Adams, for offering so much helpful advice and support over the years, both during official committee meetings and on many other occasions. I thank all the Ray lab and Dahanukar lab members for making the laboratory a fun and productive place to work. In particular, Paulina Ngo performed most of the behavior tests reported in Chapter 4, Tom Guda helped with many behavioral assays, and Greg Pask helped with cloning. -

Genomic and Transcriptomic Insights Into Calcium Carbonate Biomineralization by Marine Actinobacterium Brevibacterium Linens BS258
fmicb-08-00602 April 4, 2017 Time: 15:2 # 1 ORIGINAL RESEARCH published: 06 April 2017 doi: 10.3389/fmicb.2017.00602 Genomic and Transcriptomic Insights into Calcium Carbonate Biomineralization by Marine Actinobacterium Brevibacterium linens BS258 Yuying Zhu1,2,3†, Ning Ma1,2,3†, Weihua Jin4, Shimei Wu5 and Chaomin Sun1,2* 1 Key Laboratory of Experimental Marine Biology, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China, 2 Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China, 3 College of Earth Science, University of Chinese Academy of Sciences, Beijing, China, 4 College of Biotechnology and Bioengineering, Zhejiang University of Technology, Hangzhou, China, 5 College of Life Sciences, Qingdao University, Qingdao, China Calcium carbonate (CaCO3) biomineralization has been investigated due to its wide range of scientific and technological implications, however, the molecular mechanisms of this important geomicrobiological process are largely unknown. Here, a urease- Edited by: positive marine actinobacterium Brevibacterium linens BS258 was demonstrated to Qiang Wang, Institute of Hydrobiology (CAS), China effectively form CaCO3 precipitates. Surprisingly, this bacterium could also dissolve 2C Reviewed by: the formed CaCO3 with the increase of the Ca concentration. To disclose the Xiaoke Hu, mechanisms of biomineralization, the genome of B. linens BS258 was further completely Yantai Institute of Coastal Zone Research (CAS), China sequenced. Interestingly, the expression of three carbonic anhydrases was significantly C Bruno A. Martinez, up-regulated along with the increase of Ca2 concentration and the extent of calcite Lawrence Berkeley National dissolution. Moreover, transcriptome analyses revealed that increasing concentration Laboratory, USA of Ca2C induced KEGG pathways including quorum sensing (QS) in B. -

Rind Austrian Hard Cheese Surfaces and Its Production Environment
Animal Science Publications Animal Science 2-2018 Autochthonous facility-specific microbiota dominates washed- rind Austrian hard cheese surfaces and its production environment Narciso M. Quijada University of Veterinary Medicine Vienna Evelyne Mann University of Veterinary Medicine Vienna Martin Wagner University of Veterinary Medicine Vienna David Rodríguez-Lázaro Universidad de Burgos Marta Hernández Instituto Tecnológico Agrario de Castilla y León See next page for additional authors Follow this and additional works at: https://lib.dr.iastate.edu/ans_pubs Part of the Bacteriology Commons, Environmental Microbiology and Microbial Ecology Commons, Food Processing Commons, and the Genetics and Genomics Commons The complete bibliographic information for this item can be found at https://lib.dr.iastate.edu/ ans_pubs/525. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Animal Science at Iowa State University Digital Repository. It has been accepted for inclusion in Animal Science Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Autochthonous facility-specific microbiota dominates washed-rind Austrian hard cheese surfaces and its production environment Abstract Cheese ripening involves the succession of complex microbial communities that are responsible for the organoleptic properties of the final products. The food processing environment can act as a source of natural microbial inoculation, especially in traditionally manufactured products. Austrian Vorarlberger Bergkäse (VB) is an artisanal washed-rind hard cheese produced in the western part of Austria without the addition of external ripening cultures. -

Brevibacterium from Austrian Hard Cheese Harbor a Putative Histamine Catabolism Pathway and a Plasmid for Adaptation to the Cheese Environment
Animal Science Publications Animal Science 2019 Brevibacterium from Austrian hard cheese harbor a putative histamine catabolism pathway and a plasmid for adaptation to the cheese environment Justin M. Anast Iowa State University, [email protected] Monika Dzieciol University of Veterinary Medicine Vienna Dylan L. Schultz Iowa State University, [email protected] Martin Wagner University of Veterinary Medicine Vienna Evelyne Mann University of Veterinary Medicine Vienna See next page for additional authors Follow this and additional works at: https://lib.dr.iastate.edu/ans_pubs Part of the Agriculture Commons, Animal Sciences Commons, Food Microbiology Commons, and the Genetics and Genomics Commons The complete bibliographic information for this item can be found at https://lib.dr.iastate.edu/ ans_pubs/520. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Animal Science at Iowa State University Digital Repository. It has been accepted for inclusion in Animal Science Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Brevibacterium from Austrian hard cheese harbor a putative histamine catabolism pathway and a plasmid for adaptation to the cheese environment Abstract The genus Brevibacterium harbors many members important for cheese ripening. We performed real-time quantitative PCR (qPCR) to determine the abundance of Brevibacterium on rinds of Vorarlberger Bergkäse, an Austrian artisanal washed-rind hard cheese, over 160 days of ripening. Our results show that Brevibacterium are abundant on Vorarlberger Bergkäse rinds throughout the ripening time. -
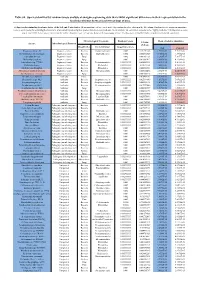
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp. -

Microbial Interactions Within the Cheese Ecosystem and Their Application to Improve Quality and Safety
foods Review Microbial Interactions within the Cheese Ecosystem and Their Application to Improve Quality and Safety Baltasar Mayo * , Javier Rodríguez, Lucía Vázquez and Ana Belén Flórez Departamento de Microbiología y Bioquímica, Instituto de Productos Lácteos de Asturias (IPLA), Consejo Superior de Investigaciones Científicas (CSIC), Paseo Río Linares s/n, 33300 Villaviciosa, Spain; [email protected] (J.R.); [email protected] (L.V.); abfl[email protected] (A.B.F.) * Correspondence: [email protected]; Tel.: +34-985893345 Abstract: The cheese microbiota comprises a consortium of prokaryotic, eukaryotic and viral popula- tions, among which lactic acid bacteria (LAB) are majority components with a prominent role during manufacturing and ripening. The assortment, numbers and proportions of LAB and other microbial biotypes making up the microbiota of cheese are affected by a range of biotic and abiotic factors. Cooperative and competitive interactions between distinct members of the microbiota may occur, with rheological, organoleptic and safety implications for ripened cheese. However, the mechanistic details of these interactions, and their functional consequences, are largely unknown. Acquiring such knowledge is important if we are to predict when fermentations will be successful and understand the causes of technological failures. The experimental use of “synthetic” microbial communities might help throw light on the dynamics of different cheese microbiota components and the interplay Citation: Mayo, B.; Rodríguez, J.; between them. Although synthetic communities cannot reproduce entirely the natural microbial di- Vázquez, L.; Flórez, A.B. Microbial versity in cheese, they could help reveal basic principles governing the interactions between microbial Interactions within the Cheese types and perhaps allow multi-species microbial communities to be developed as functional starters. -

Comparative Genomic Analysis of Brevibacterium Strains
Pham et al. BMC Genomics (2017) 18:955 DOI 10.1186/s12864-017-4322-1 RESEARCHARTICLE Open Access Comparative genomic analysis of Brevibacterium strains: insights into key genetic determinants involved in adaptation to the cheese habitat Nguyen-Phuong Pham1, Séverine Layec1, Eric Dugat-Bony1, Marie Vidal2, Françoise Irlinger1 and Christophe Monnet1* Abstract Background: Brevibacterium strains are widely used for the manufacturing of surface-ripened cheeses, contributing to the breakdown of lipids and proteins and producing volatile sulfur compounds and red-orange pigments. The objective of the present study was to perform comparative genomic analyses in order to better understand the mechanisms involved in their ability to grow on the cheese surface and the differences between the strains. Results: The genomes of 23 Brevibacterium strains, including twelve strains isolated from cheeses, were compared for their gene repertoire involved in salt tolerance, iron acquisition, bacteriocin production and the ability to use the energy compounds present in cheeses. All or almost all the genomes encode the enzymes involved in ethanol, acetate, lactate, 4-aminobutyrate and glycerol catabolism, and in the synthesis of the osmoprotectants ectoine, glycine-betaine and trehalose. Most of the genomes contain two contiguous genes encoding extracellular proteases, one of which was previously characterized for its activity on caseins. Genes encoding a secreted triacylglycerol lipase or involved in the catabolism of galactose and D-galactonate or in the synthesis of a hydroxamate-type siderophore are present in part of the genomes. Numerous Fe3+/siderophore ABC transport components are present, part of them resulting from horizontal gene transfers. Two cheese-associated strains have also acquired catecholate-type siderophore biosynthesis gene clusters by horizontal gene transfer. -

Homebrew Cheese Making for Beer Lovers HOME BREW CON 2018 Similarities
Homebrew Cheese making for Beer Lovers HOME BREW CON 2018 Similarities • The science behind the transformation from plants to milk to cheese is amazing. • In fact, cheese has much in common with wine and beer: – They result from fermentation by microorganisms; they are “value-added” products where processing greatly increases the value; and they reflect local climate and terrain. • Cheese has fascinated humanity for a long time, inspiring people to refer to it as everything from “the wine of foods” (Vivenne Marquis and Patricia Haskell) to “milk’s leap toward immortality” (Clifton Fadiman)… Cheese • Coagulation of the milk protein casein – Acidification followed by some form of rennet • Mostly proteins and fat as a result • Typically cow, goat, sheep or buffalo • Solids separated typically into a mold • Pressing makes for a a harder, denser cheese A bit of Science Cow Milk Component Approximate % Water 86.5 Lactose 4.8 Fat 4.5 Proteins 3.5 Vitamins and Minerals 0.7 Milk is an emulsion of fat globules and a suspension of casein micelles. These are suspended in the liquid phase of milk that contains dissolved lactose, whey proteins and some minerals. The Milk • Quality ingredients produce better end results – get the best milk you can afford – RAW – comes with its own culture – Grass Fed – healthier balance of Omega 3/6 – Organic – control what goes into it – Biodynamic – sustainable + soil health – A2 – may be more tolerable to some folks – Whole – depending on the cheese Sourcing • Get to know your milk supplier • Ask about their processes -

Bacterial Community Assembly from Cow Teat Skin to Ripened Cheeses Is
www.nature.com/scientificreports OPEN Bacterial community assembly from cow teat skin to ripened cheeses is infuenced by grazing Received: 15 September 2017 Accepted: 11 December 2017 systems Published: xx xx xxxx Marie Frétin1,2, Bruno Martin2, Etienne Rifa 1, Verdier-Metz Isabelle1, Dominique Pomiès2, Anne Ferlay2, Marie-Christine Montel1 & Céline Delbès 1 The objectives of this study were to explore bacterial community assembly from cow teat skin to raw milk cheeses and to evaluate the role of farming systems on this assembly using 16S rRNA gene high-throughput sequencing. The two grazing systems studied (extensive vs. semi-extensive) had a greater efect on the microbiota of cow teat skin than on that of raw milks and cheeses. On teat skin, the relative abundance of several taxa at diferent taxonomic levels (Coriobacteriia, Bifdobacteriales, Corynebacteriales, Lachnospiraceae, Atopobium, and Clostridium) varied depending on the grazing system and the period (early or late summer). In cheese, the abundance of sub-dominant lactic acid bacteria (LAB) varied depending on the grazing system. Overall, 85% of OTUs detected in raw milks and 27% of OTUs detected in ripened cheeses were also found on cow teat skin. Several shared OTUs were assigned to taxa known to be involved in the development of cheese sensory characteristics, such as Micrococcales, Staphylococcaceae, and LAB. Our results highlight the key role of cow teat skin as a reservoir of microbial diversity for raw milk, and for the frst time, that cow teat skin serves as a potential source of microorganisms found in raw-milk cheeses. Te microbial community of cow teat skin depends on the dairy farm environment1. -

New Antimicrobials to Target Gut and Food Pathogens
New antimicrobials to target gut and food pathogens Enriqueta Garcia-Gutierrez A thesis submitted for the degree of Doctor of Philosophy to the University of East Anglia Quadram Institute Bioscience Teagasc September 2019 ©This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognise that its copyright rests with the author and that use of any information derived therefrom must be in accordance with current UK Copyright Law. In addition, any quotation or extract must include full attribution. PhD Thesis 2019 Enriqueta Garcia-Gutierrez New Antimicrobials to Target Gut and Food Pathogens ABSTRACT There is a pressing need for the discovery of new antimicrobials to fight antibiotic resistant bacteria. The aim of this thesis was the discovery and characterisation of new bacteriocins from two sources, fermented foods and human faeces, testing the hypothesis that bacteria from the same niche will produce antimicrobials uniquely suited to act in this niche. Isolates from culture collections and new isolates from food and faecal samples were screened against a panel of pathogens responsible for food spoilage and human disease. Promising candidates were selected for genome sequencing, antimicrobial characterisation and biological study. The genome of Lactobacillus gasseri LM19 showed the presence of antimicrobial genes encoding, among others, a new bacteriocin, gassericin M. L. gasseri LM19 could survive and express its bacteriocin genes under colonic conditions. Its administration modulated the effects of Clostridium perfringens on the gut microbiota composition. Streptococcus agalactiae DPC7040 was previously shown to produce the natural variant nisin P. MALDI-ToF analysis confirmed that nisin P is three amino acids shorter than nisin A and that two lanthionine rings were absent in 50% of molecules.