Download (15MB)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Data
Figure 2S 4 7 A - C 080125 CSCs 080418 CSCs - + IFN-a 48 h + IFN-a 48 h + IFN-a 72 h 6 + IFN-a 72 h 3 5 MRFI 4 2 3 2 1 1 0 0 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 7 B 13 080125 FBS - D 080418 FBS - + IFN-a 48 h 12 + IFN-a 48 h + IFN-a 72 h + IFN-a 72 h 6 080125 FBS 11 10 5 9 8 4 7 6 3 MRFI 5 4 2 3 2 1 1 0 0 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 Molecule Molecule FIGURE 4S FIGURE 5S Panel A Panel B FIGURE 6S A B C D Supplemental Results Table 1S. Modulation by IFN-α of APM in GBM CSC and FBS tumor cell lines. Molecule * Cell line IFN-α‡ HLA β2-m# HLA LMP TAP1 TAP2 class II A A HC§ 2 7 10 080125 CSCs - 1∞ (1) 3 (65) 2 (91) 1 (2) 6 (47) 2 (61) 1 (3) 1 (2) 1 (3) + 2 (81) 11 (80) 13 (99) 1 (3) 8 (88) 4 (91) 1 (2) 1 (3) 2 (68) 080125 FBS - 2 (81) 4 (63) 4 (83) 1 (3) 6 (80) 3 (67) 2 (86) 1 (3) 2 (75) + 2 (99) 14 (90) 7 (97) 5 (75) 7 (100) 6 (98) 2 (90) 1 (4) 3 (87) 080418 CSCs - 2 (51) 1 (1) 1 (3) 2 (47) 2 (83) 2 (54) 1 (4) 1 (2) 1 (3) + 2 (81) 3 (76) 5 (75) 2 (50) 2 (83) 3 (71) 1 (3) 2 (87) 1 (2) 080418 FBS - 1 (3) 3 (70) 2 (88) 1 (4) 3 (87) 2 (76) 1 (3) 1 (3) 1 (2) + 2 (78) 7 (98) 5 (99) 2 (94) 5 (100) 3 (100) 1 (4) 2 (100) 1 (2) 070104 CSCs - 1 (2) 1 (3) 1 (3) 2 (78) 1 (3) 1 (2) 1 (3) 1 (3) 1 (2) + 2 (98) 8 (100) 10 (88) 4 (89) 3 (98) 3 (94) 1 (4) 2 (86) 2 (79) * expression of APM molecules was evaluated by intracellular staining and cytofluorimetric analysis; ‡ cells were treatead or not (+/-) for 72 h with 1000 IU/ml of IFN-α; # β-2 microglobulin; § β-2 microglobulin-free HLA-A heavy chain; ∞ values are indicated as ratio between the mean of fluorescence intensity of cells stained with the selected mAb and that of the negative control; bold values indicate significant MRFI (≥ 2). -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Overexpression of Salicylic Acid Carboxyl Methyltransferase (Cssamt1) Enhances Tolerance to Huanglongbing Disease in Wanjincheng Orange (Citrus Sinensis (L.) Osbeck)
International Journal of Molecular Sciences Article Overexpression of Salicylic Acid Carboxyl Methyltransferase (CsSAMT1) Enhances Tolerance to Huanglongbing Disease in Wanjincheng Orange (Citrus sinensis (L.) Osbeck) Xiuping Zou * , Ke Zhao, Yunuo Liu, Meixia Du, Lin Zheng, Shuai Wang, Lanzhen Xu, Aihong Peng, Yongrui He, Qin Long and Shanchun Chen * Citrus Research Institute, Southwest University/Chinese Academy of Agricultural Sciences, Chongqing 400716, China; [email protected] (K.Z.); [email protected] (Y.L.); [email protected] (M.D.); [email protected] (L.Z.); [email protected] (S.W.); [email protected] (L.X.); [email protected] (A.P.); [email protected] (Y.H.); [email protected] (Q.L.) * Correspondence: [email protected] (X.Z.); [email protected] (S.C.) Abstract: Citrus Huanglongbing (HLB) disease or citrus greening is caused by Candidatus Liberibacter asiaticus (Las) and is the most devastating disease in the global citrus industry. Salicylic acid (SA) plays a central role in regulating plant defenses against pathogenic attack. SA methyltransferase (SAMT) modulates SA homeostasis by converting SA to methyl salicylate (MeSA). Here, we report on the functions of the citrus SAMT (CsSAMT1) gene from HLB-susceptible Wanjincheng orange Citation: Zou, X.; Zhao, K.; Liu, Y.; (Citrus sinensis (L.) Osbeck) in plant defenses against Las infection. The CsSAMT1 cDNA was Du, M.; Zheng, L.; Wang, S.; Xu, L.; expressed in yeast. Using in vitro enzyme assays, yeast expressing CsSAMT1 was confirmed to Peng, A.; He, Y.; Long, Q.; et al. specifically catalyze the formation of MeSA using SA as a substrate. Transgenic Wanjincheng orange Overexpression of Salicylic Acid plants overexpressing CsSAMT1 had significantly increased levels of SA and MeSA compared to Carboxyl Methyltransferase wild-type controls. -

The Direction of Cross Affects Obesity After Puberty in Male but Not Female
Kärst et al. BMC Genomics (2015) 16:904 DOI 10.1186/s12864-015-2164-2 RESEARCH ARTICLE Open Access The direction of cross affects obesity after puberty in male but not female offspring Stefan Kärst1†, Danny Arends1†, Sebastian Heise1†, Jan Trost1, Marie-Laure Yaspo2, Vyacheslav Amstislavskiy2, Thomas Risch2, Hans Lehrach2 and Gudrun A. Brockmann1* Abstract Background: We investigated parent-of-origin and allele-specific expression effects on obesity and hepatic gene expression in reciprocal crosses between the Berlin Fat Mouse Inbred line (BFMI) and C57Bl/6NCrl (B6N). Results: We found that F1-males with a BFMI mother developed 1.8 times more fat mass on a high fat diet at 10 weeks than F1-males of a BFMI father. The phenotype was detectable from six weeks on and was preserved after cross-fostering. RNA-seq data of liver provided evidence for higher biosynthesis and elongation of fatty acids (p = 0.00635) in obese male offspring of a BFMI mother versus lean offspring of a BFMI father. Furthermore, fatty acid degradation (p = 0.00198) and the peroxisome pathway were impaired (p = 0.00094). The circadian rhythm was affected as well (p = 0.00087). Among the highest up-regulated protein coding genes in obese males were Acot4 (1.82 fold, p = 0.022), Cyp4a10 (1.35 fold, p = 0.026) and Cyp4a14 (1.32 fold, p = 0.012), which hydroxylize fatty acids and which are known to be increased in liver steatosis. Obese males showed lower expression of the genetically imprinted and paternally expressed 3 (Peg3) gene (0.31 fold, p = 0.046) and higher expression of the androgen receptor (Ar) gene (2.38 fold, p = 0.068). -

Investigation of Candidate Genes and Mechanisms Underlying Obesity
Prashanth et al. BMC Endocrine Disorders (2021) 21:80 https://doi.org/10.1186/s12902-021-00718-5 RESEARCH ARTICLE Open Access Investigation of candidate genes and mechanisms underlying obesity associated type 2 diabetes mellitus using bioinformatics analysis and screening of small drug molecules G. Prashanth1 , Basavaraj Vastrad2 , Anandkumar Tengli3 , Chanabasayya Vastrad4* and Iranna Kotturshetti5 Abstract Background: Obesity associated type 2 diabetes mellitus is a metabolic disorder ; however, the etiology of obesity associated type 2 diabetes mellitus remains largely unknown. There is an urgent need to further broaden the understanding of the molecular mechanism associated in obesity associated type 2 diabetes mellitus. Methods: To screen the differentially expressed genes (DEGs) that might play essential roles in obesity associated type 2 diabetes mellitus, the publicly available expression profiling by high throughput sequencing data (GSE143319) was downloaded and screened for DEGs. Then, Gene Ontology (GO) and REACTOME pathway enrichment analysis were performed. The protein - protein interaction network, miRNA - target genes regulatory network and TF-target gene regulatory network were constructed and analyzed for identification of hub and target genes. The hub genes were validated by receiver operating characteristic (ROC) curve analysis and RT- PCR analysis. Finally, a molecular docking study was performed on over expressed proteins to predict the target small drug molecules. Results: A total of 820 DEGs were identified between -

Loss of Fam60a, a Sin3a Subunit, Results in Embryonic Lethality and Is Associated with Aberrant Methylation at a Subset of Gene
RESEARCH ARTICLE Loss of Fam60a, a Sin3a subunit, results in embryonic lethality and is associated with aberrant methylation at a subset of gene promoters Ryo Nabeshima1,2, Osamu Nishimura3,4, Takako Maeda1, Natsumi Shimizu2, Takahiro Ide2, Kenta Yashiro1†, Yasuo Sakai1, Chikara Meno1, Mitsutaka Kadota3,4, Hidetaka Shiratori1†, Shigehiro Kuraku3,4*, Hiroshi Hamada1,2* 1Developmental Genetics Group, Graduate School of Frontier Biosciences, Osaka University, Suita, Japan; 2Laboratory for Organismal Patterning, RIKEN Center for Developmental Biology, Kobe, Japan; 3Phyloinformatics Unit, RIKEN Center for Life Science Technologies, Kobe, Japan; 4Laboratory for Phyloinformatics, RIKEN Center for Biosystems Dynamics Research, Kobe, Japan Abstract We have examined the role of Fam60a, a gene highly expressed in embryonic stem cells, in mouse development. Fam60a interacts with components of the Sin3a-Hdac transcriptional corepressor complex, and most Fam60a–/– embryos manifest hypoplasia of visceral organs and die in utero. Fam60a is recruited to the promoter regions of a subset of genes, with the expression of these genes being either up- or down-regulated in Fam60a–/– embryos. The DNA methylation level of the Fam60a target gene Adhfe1 is maintained at embryonic day (E) 7.5 but markedly reduced at –/– *For correspondence: E9.5 in Fam60a embryos, suggesting that DNA demethylation is enhanced in the mutant. [email protected] (SK); Examination of genome-wide DNA methylation identified several differentially methylated regions, [email protected] (HH) which were preferentially hypomethylated, in Fam60a–/– embryos. Our data suggest that Fam60a is †These authors contributed required for proper embryogenesis, at least in part as a result of its regulation of DNA methylation equally to this work at specific gene promoters. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

Ten Commandments for a Good Scientist
Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds Ana María Sotoca Covaleda Wageningen 2010 Thesis committee Thesis supervisors Prof. dr. ir. Ivonne M.C.M. Rietjens Professor of Toxicology Wageningen University Prof. dr. Albertinka J. Murk Personal chair at the sub-department of Toxicology Wageningen University Thesis co-supervisor Dr. ir. Jacques J.M. Vervoort Associate professor at the Laboratory of Biochemistry Wageningen University Other members Prof. dr. Michael R. Muller, Wageningen University Prof. dr. ir. Huub F.J. Savelkoul, Wageningen University Prof. dr. Everardus J. van Zoelen, Radboud University Nijmegen Dr. ir. Toine F.H. Bovee, RIKILT, Wageningen This research was conducted under the auspices of the Graduate School VLAG Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds Ana María Sotoca Covaleda Thesis submitted in fulfillment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. dr. M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Tuesday 14 September 2010 at 4 p.m. in the Aula Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds. Ana María Sotoca Covaleda Thesis Wageningen University, Wageningen, The Netherlands, 2010, With references, and with summary in Dutch. ISBN: 978-90-8585-707-5 “Caminante no hay camino, se hace camino al andar. Al andar se hace camino, y al volver la vista atrás se ve la senda que nunca se ha de volver a pisar” - Antonio Machado – A mi madre. -

(12) Patent Application Publication (10) Pub. No.: US 2006/0088532 A1 Alitalo Et Al
US 20060O88532A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2006/0088532 A1 Alitalo et al. (43) Pub. Date: Apr. 27, 2006 (54) LYMPHATIC AND BLOOD ENDOTHELIAL Related U.S. Application Data CELL GENES (60) Provisional application No. 60/363,019, filed on Mar. (76) Inventors: Kari Alitalo, Helsinki (FI); Taija 7, 2002. Makinen, Helsinki (FI); Tatiana Petrova, Helsinki (FI); Pipsa Publication Classification Saharinen, Helsinki (FI); Juha Saharinen, Helsinki (FI) (51) Int. Cl. A6IR 48/00 (2006.01) Correspondence Address: A 6LX 39/395 (2006.01) MARSHALL, GERSTEIN & BORUN LLP A6II 38/18 (2006.01) 233 S. WACKER DRIVE, SUITE 6300 (52) U.S. Cl. .............................. 424/145.1: 514/2: 514/44 SEARS TOWER (57) ABSTRACT CHICAGO, IL 60606 (US) The invention provides polynucleotides and genes that are (21) Appl. No.: 10/505,928 differentially expressed in lymphatic versus blood vascular endothelial cells. These genes are useful for treating diseases (22) PCT Filed: Mar. 7, 2003 involving lymphatic vessels, such as lymphedema, various inflammatory diseases, and cancer metastasis via the lym (86). PCT No.: PCT/USO3FO6900 phatic system. Patent Application Publication Apr. 27, 2006 Sheet 1 of 2 US 2006/0088532 A1 integrin O9 integrin O1 KIAAO711 KAAO644 ApoD Fig. 1 Patent Application Publication Apr. 27, 2006 Sheet 2 of 2 US 2006/0088532 A1 CN g uueleo-gº US 2006/0O88532 A1 Apr. 27, 2006 LYMPHATIC AND BLOOD ENDOTHELLAL CELL lymphatic vessels, such as lymphangiomas or lymphang GENES iectasis. Witte, et al., Regulation of Angiogenesis (eds. Goldber, I. D. & Rosen, E. M.) 65-112 (Birkauser, Basel, BACKGROUND OF THE INVENTION Switzerland, 1997). -

(BPA) Exposure Biomarkers in Ovarian Cancer
Journal of Clinical Medicine Article Identification of Potential Bisphenol A (BPA) Exposure Biomarkers in Ovarian Cancer Aeman Zahra 1, Qiduo Dong 1, Marcia Hall 1,2 , Jeyarooban Jeyaneethi 1, Elisabete Silva 1, Emmanouil Karteris 1,* and Cristina Sisu 1,* 1 Biosciences, College of Health, Medicine and Life Sciences, Brunel University London, Uxbridge UB8 3PH, UK; [email protected] (A.Z.); [email protected] (Q.D.); [email protected] (M.H.); [email protected] (J.J.); [email protected] (E.S.) 2 Mount Vernon Cancer Centre, Northwood HA6 2RN, UK * Correspondence: [email protected] (E.K.); [email protected] (C.S.) Abstract: Endocrine-disrupting chemicals (EDCs) can exert multiple deleterious effects and have been implicated in carcinogenesis. The xenoestrogen Bisphenol A (BPA) that is found in various consumer products has been involved in the dysregulation of numerous signalling pathways. In this paper, we present the analysis of a set of 94 genes that have been shown to be dysregulated in presence of BPA in ovarian cancer cell lines since we hypothesised that these genes might be of biomarker potential. This study sought to identify biomarkers of disease and biomarkers of disease- associated exposure. In silico analyses took place using gene expression data extracted from The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression (GTEx) databases. Differential expression was further validated at protein level using immunohistochemistry on an ovarian cancer tissue microarray. We found that 14 out of 94 genes are solely dysregulated in the presence of BPA, while the remaining 80 genes are already dysregulated (p-value < 0.05) in their expression pattern Citation: Zahra, A.; Dong, Q.; Hall, as a consequence of the disease. -
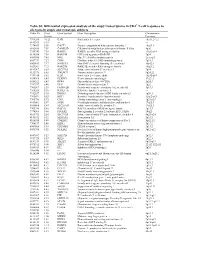
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3 -

140503 IPF Signatures Supplement Withfigs Thorax
Supplementary material for Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis Daryle J. DePianto1*, Sanjay Chandriani1⌘*, Alexander R. Abbas1, Guiquan Jia1, Elsa N. N’Diaye1, Patrick Caplazi1, Steven E. Kauder1, Sabyasachi Biswas1, Satyajit K. Karnik1#, Connie Ha1, Zora Modrusan1, Michael A. Matthay2, Jasleen Kukreja3, Harold R. Collard2, Jackson G. Egen1, Paul J. Wolters2§, and Joseph R. Arron1§ 1Genentech Research and Early Development, South San Francisco, CA 2Department of Medicine, University of California, San Francisco, CA 3Department of Surgery, University of California, San Francisco, CA ⌘Current address: Novartis Institutes for Biomedical Research, Emeryville, CA. #Current address: Gilead Sciences, Foster City, CA. *DJD and SC contributed equally to this manuscript §PJW and JRA co-directed this project Address correspondence to Paul J. Wolters, MD University of California, San Francisco Department of Medicine Box 0111 San Francisco, CA 94143-0111 [email protected] or Joseph R. Arron, MD, PhD Genentech, Inc. MS 231C 1 DNA Way South San Francisco, CA 94080 [email protected] 1 METHODS Human lung tissue samples Tissues were obtained at UCSF from clinical samples from IPF patients at the time of biopsy or lung transplantation. All patients were seen at UCSF and the diagnosis of IPF was established through multidisciplinary review of clinical, radiological, and pathological data according to criteria established by the consensus classification of the American Thoracic Society (ATS) and European Respiratory Society (ERS), Japanese Respiratory Society (JRS), and the Latin American Thoracic Association (ALAT) (ref. 5 in main text). Non-diseased normal lung tissues were procured from lungs not used by the Northern California Transplant Donor Network.