The Solvation Interface Is a Determining Factor in Peptide Conformational Preferences
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Network of Molecular Switches Controls the Activation of the Two-Component Response Regulator Ntrc
ARTICLE Received 8 Aug 2014 | Accepted 26 Apr 2015 | Published 15 Jun 2015 DOI: 10.1038/ncomms8283 A network of molecular switches controls the activation of the two-component response regulator NtrC Dan K. Vanatta1, Diwakar Shukla1,2,3, Morgan Lawrenz1 & Vijay S. Pande1,2 Recent successes in simulating protein structure and folding dynamics have demonstrated the power of molecular dynamics to predict the long timescale behaviour of proteins. Here, we extend and improve these methods to predict molecular switches that characterize conformational change pathways between the active and inactive state of nitrogen regulatory protein C (NtrC). By employing unbiased Markov state model-based molecular dynamics simulations, we construct a dynamic picture of the activation pathways of this key bacterial signalling protein that is consistent with experimental observations and predicts new mutants that could be used for validation of the mechanism. Moreover, these results suggest a novel mechanistic paradigm for conformational switching. 1 Department of Chemistry, Stanford University, Stanford, California 94305, USA. 2 SIMBIOS NIH Center for Biomedical Computation, Stanford University, Stanford, California 94305,USA. 3 Department of Chemical and Bimolecular Engineering, University of Illinois at Urbana-Champaign, Urbana, Illionios 61801, USA. Correspondence and requests for materials should be addressed to V.S.P. (email: [email protected]). NATURE COMMUNICATIONS | 6:7283 | DOI: 10.1038/ncomms8283 | www.nature.com/naturecommunications 1 & 2015 Macmillan Publishers Limited. All rights reserved. ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/ncomms8283 roteins involved in cellular signalling change their con- Results formation in response to changes in environment (input Simulations reveal molecular switches for NtrC activation. Psignal), such as ligand binding or chemical modification, MSMs for NtrC were built using snapshots along the simulated to control downstream cellular processes (output signal)1. -

Spring 2013 Lecture 23
CHM333 LECTURES 23: 3/25/13 SPRING 2013 Professor Christine Hrycyna LIPIDS III EFFECT OF CHOLESTEROL ON MEMBRANES: - Bulky rigid molecule - Moderates fluidity of membranes – both increases and decreases o Cholesterol in membranes DECREASES fluidity because it is rigid o Prevents crystallization (making solid) of fatty acyl side chains by fitting between them. Disrupts close packing of fatty acyl chains. Therefore, INCREASED fluidity BIOLOGICAL MEMBRANES CONTAIN PROTEINS AS WELL AS LIPIDS: - Proteins are 20-80% of cell membrane - Rest is lipid or carbohydrate; supramolecular assembly of lipid, protein and carbohydrate - Proteins are also distributed asymmetrically - TWO classes of Membrane Proteins: o Integral Membrane Proteins o Peripheral Membrane Proteins 178 CHM333 LECTURES 23: 3/25/13 SPRING 2013 Professor Christine Hrycyna - INTEGRAL MEMBRANE PROTEINS o Located WITHIN the lipid bilayer o Usually span the bilayer one or more times – called transmembrane (TM) proteins o Hydrophobic amino acids interact with fatty acid chains in the hydrophobic core of the membrane o Can be removed from the membrane with detergents like SDS – need to disrupt the hydrophobic interactions § Membrane Disruption Animation: o http://www.youtube.com/watch?v=AHT37pvcjc0 o Function: § Transporters – moving molecules into or out of cells or cell membranes § Receptors – transmitting signals from outside of the cell to the inside - β Barrel Integral Membrane Proteins § Barrel-shaped membrane protein that is made up of antiparallel β-strands with hydrophilic (interior) and hydrophobic (facing lipid tails). § So far found only in outer membranes of Gram-negative bacteria, cell wall of Gram-positive bacteria, and outer membranes of mitochondria and chloroplasts. 179 CHM333 LECTURES 23: 3/25/13 SPRING 2013 Professor Christine Hrycyna - α-Helical Membrane Proteins - Can cross the membrane once or many times and have multiple transmembrane segments. -

POLYMER STRUCTURE and CHARACTERIZATION Professor
POLYMER STRUCTURE AND CHARACTERIZATION Professor John A. Nairn Fall 2007 TABLE OF CONTENTS 1 INTRODUCTION 1 1.1 Definitions of Terms . 2 1.2 Course Goals . 5 2 POLYMER MOLECULAR WEIGHT 7 2.1 Introduction . 7 2.2 Number Average Molecular Weight . 9 2.3 Weight Average Molecular Weight . 10 2.4 Other Average Molecular Weights . 10 2.5 A Distribution of Molecular Weights . 11 2.6 Most Probable Molecular Weight Distribution . 12 3 MOLECULAR CONFORMATIONS 21 3.1 Introduction . 21 3.2 Nomenclature . 23 3.3 Property Calculation . 25 3.4 Freely-Jointed Chain . 27 3.4.1 Freely-Jointed Chain Analysis . 28 3.4.2 Comment on Freely-Jointed Chain . 34 3.5 Equivalent Freely Jointed Chain . 37 3.6 Vector Analysis of Polymer Conformations . 38 3.7 Freely-Rotating Chain . 41 3.8 Hindered Rotating Chain . 43 3.9 More Realistic Analysis . 45 3.10 Theta (Θ) Temperature . 47 3.11 Rotational Isomeric State Model . 48 4 RUBBER ELASTICITY 57 4.1 Introduction . 57 4.2 Historical Observations . 57 4.3 Thermodynamics . 60 4.4 Mechanical Properties . 62 4.5 Making Elastomers . 68 4.5.1 Diene Elastomers . 68 0 4.5.2 Nondiene Elastomers . 69 4.5.3 Thermoplastic Elastomers . 70 5 AMORPHOUS POLYMERS 73 5.1 Introduction . 73 5.2 The Glass Transition . 73 5.3 Free Volume Theory . 73 5.4 Physical Aging . 73 6 SEMICRYSTALLINE POLYMERS 75 6.1 Introduction . 75 6.2 Degree of Crystallization . 75 6.3 Structures . 75 Chapter 1 INTRODUCTION The topic of polymer structure and characterization covers molecular structure of polymer molecules, the arrangement of polymer molecules within a bulk polymer material, and techniques used to give information about structure or properties of polymers. -

Ideal Chain Conformations and Statistics
ENAS 606 : Polymer Physics Chinedum Osuji 01.24.2013:HO2 Ideal Chain Conformations and Statistics 1 Overview Consideration of the structure of macromolecules starts with a look at the details of chain level chemical details which can impact the conformations adopted by the polymer. In the case of saturated carbon ◦ backbones, while maintaining the desired Ci−1 − Ci − Ci+1 bond angle of 112 , the placement of the final carbon in the triad above can occur at any point along the circumference of a circle, defining a torsion angle '. We can readily recognize the energetic differences as a function this angle, U(') such that there are 3 minima - a deep minimum corresponding the the trans state, for which ' = 0 and energetically equivalent gauche− and gauche+ states at ' = ±120 degrees, as shown in Fig 1. Figure 1: Trans, gauche- and gauche+ configurations, and their energetic sates 1.1 Static Flexibility The static flexibility of the chain in equilibrium is determined by the difference between the levels of the energy minima corresponding the gauche and trans states, ∆. If ∆ < kT , the g+, g− and t states occur with similar probability, and so the chain can change direction and appears as a random coil. If ∆ takes on a larger value, then the t conformations will be enriched, so the chain will be rigid locally, but on larger length scales, the eventual occurrence of g+ and g− conformations imparts a random conformation. Overall, if we ignore details on some length scale smaller than lp, the persistence length, the polymer appears as a continuous flexible chain where 1 lp = l0 exp(∆/kT ) (1) where l0 is something like a monomer length. -

Site-Specific Tryptophan Labels Reveal Local Microsecond
molecules Article Site-Specific Tryptophan Labels Reveal Local Microsecond–Millisecond Motions of Dihydrofolate Reductase Morgan B. Vaughn 1,*, Chloe Biren 2, Qun Li 2, Ashwin Ragupathi 2 and R. Brian Dyer 2,* 1 Division of Natural Sciences, Oglethorpe University, Atlanta, GA 30319, USA 2 Department of Chemistry, Emory University, Atlanta, GA 30322, USA; [email protected] (C.B.); [email protected] (Q.L.); [email protected] (A.R.) * Correspondence: [email protected] (M.B.V.); [email protected] (R.B.D.); Tel.: +1-404-504-1214 (M.B.V.); +1-404-727-6637 (R.B.D.) Academic Editor: Rafik Karaman Received: 24 July 2020; Accepted: 21 August 2020; Published: 22 August 2020 Abstract: Many enzymes are known to change conformations during their catalytic cycle, but the role of these protein motions is not well understood. Escherichia coli dihydrofolate reductase (DHFR) is a small, flexible enzyme that is often used as a model system for understanding enzyme dynamics. Recently, native tryptophan fluorescence was used as a probe to study micro- to millisecond dynamics of DHFR. Yet, because DHFR has five native tryptophans, the origin of the observed conformational changes could not be assigned to a specific region within the enzyme. Here, we use DHFR mutants, each with a single tryptophan as a probe for temperature jump fluorescence spectroscopy, to further inform our understanding of DHFR dynamics. The equilibrium tryptophan fluorescence of the mutants shows that each tryptophan is in a different environment and that wild-type DHFR fluorescence is not a simple summation of all the individual tryptophan fluorescence signatures due to tryptophan–tryptophan interactions. -

Predicting Protein-Membrane Interfaces of Peripheral Membrane
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.28.450157; this version posted June 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Predicting protein-membrane interfaces of pe- ripheral membrane proteins using ensemble machine learning Alexios Chatzigoulas1,2,* and Zoe Cournia1,* 1Biomedical Research Foundation, Academy of Athens, 4 Soranou Ephessiou, 11527 Athens, Greece, 2Depart- ment of Informatics and Telecommunications, National and Kapodistrian University of Athens, 15784 Athens, Greece *To whom correspondence should be addressed. Abstract Motivation: Abnormal protein-membrane attachment is involved in deregulated cellular pathways and in disease. Therefore, the possibility to modulate protein-membrane interactions represents a new promising therapeutic strategy for peripheral membrane proteins that have been considered so far undruggable. A major obstacle in this drug design strategy is that the membrane binding domains of peripheral membrane proteins are usually not known. The development of fast and efficient algorithms predicting the protein-membrane interface would shed light into the accessibility of membrane-protein interfaces by drug-like molecules. Results: Herein, we describe an ensemble machine learning methodology and algorithm for predicting membrane-penetrating residues. We utilize available experimental data in the literature for training 21 machine learning classifiers and a voting classifier. Evaluation of the ensemble classifier accuracy pro- duced a macro-averaged F1 score = 0.92 and an MCC = 0.84 for predicting correctly membrane-pen- etrating residues on unknown proteins of an independent test set. -

Native-Like Mean Structure in the Unfolded Ensemble of Small Proteins
B doi:10.1016/S0022-2836(02)00888-4 available online at http://www.idealibrary.com on w J. Mol. Biol. (2002) 323, 153–164 Native-like Mean Structure in the Unfolded Ensemble of Small Proteins Bojan Zagrovic1, Christopher D. Snow1, Siraj Khaliq2 Michael R. Shirts2 and Vijay S. Pande1,2* 1Biophysics Program The nature of the unfolded state plays a great role in our understanding of Stanford University, Stanford proteins. However, accurately studying the unfolded state with computer CA 94305-5080, USA simulation is difficult, due to its complexity and the great deal of sampling required. Using a supercluster of over 10,000 processors we 2Department of Chemistry have performed close to 800 ms of molecular dynamics simulation in Stanford University, Stanford atomistic detail of the folded and unfolded states of three polypeptides CA 94305-5080, USA from a range of structural classes: the all-alpha villin headpiece molecule, the beta hairpin tryptophan zipper, and a designed alpha-beta zinc finger mimic. A comparison between the folded and the unfolded ensembles reveals that, even though virtually none of the individual members of the unfolded ensemble exhibits native-like features, the mean unfolded structure (averaged over the entire unfolded ensemble) has a native-like geometry. This suggests several novel implications for protein folding and structure prediction as well as new interpretations for experiments which find structure in ensemble-averaged measurements. q 2002 Elsevier Science Ltd. All rights reserved Keywords: mean-structure hypothesis; unfolded state of proteins; *Corresponding author distributed computing; conformational averaging Introduction under folding conditions, with some notable exceptions.14 – 16 This is understandable since under Historically, the unfolded state of proteins has such conditions the unfolded state is an unstable, received significantly less attention than the folded fleeting species making any kind of quantitative state.1 The reasons for this are primarily its struc- experimental measurement very difficult. -
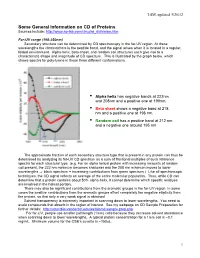
CD of Proteins Sources Include
LSM, updated 3/26/12 Some General Information on CD of Proteins Sources include: http://www.ap-lab.com/circular_dichroism.htm Far-UV range (190-250nm) Secondary structure can be determined by CD spectroscopy in the far-UV region. At these wavelengths the chromophore is the peptide bond, and the signal arises when it is located in a regular, folded environment. Alpha-helix, beta-sheet, and random coil structures each give rise to a characteristic shape and magnitude of CD spectrum. This is illustrated by the graph below, which shows spectra for poly-lysine in these three different conformations. • Alpha helix has negative bands at 222nm and 208nm and a positive one at 190nm. • Beta sheet shows a negative band at 218 nm and a positive one at 196 nm. • Random coil has a positive band at 212 nm and a negative one around 195 nm. The approximate fraction of each secondary structure type that is present in any protein can thus be determined by analyzing its far-UV CD spectrum as a sum of fractional multiples of such reference spectra for each structural type. (e.g. For an alpha helical protein with increasing amounts of random coil present, the 222 nm minimum becomes shallower and the 208 nm minimum moves to lower wavelengths ⇒ black spectrum + increasing contributions from green spectrum.) Like all spectroscopic techniques, the CD signal reflects an average of the entire molecular population. Thus, while CD can determine that a protein contains about 50% alpha-helix, it cannot determine which specific residues are involved in the helical portion. -

Conformational Proofreading: the Impact of Conformational
Conformational Proofreading: The Impact of Conformational Changes on the Specificity of Molecular Recognition Yonatan Savir1 and Tsvi Tlusty1,* 1Department of Physics of Complex Systems, the Weizmann Institute of Science, Rehovot, Israel, 76100 *Corresponding author. E-mail: [email protected] Abstract To perform recognition, molecules must locate and specifically bind their targets within a noisy biochemical environment with many look-alikes. Molecular recognition processes, especially the induced-fit mechanism, are known to involve conformational changes. This arises a basic question: does molecular recognition gain any advantage by such conformational changes? By introducing a simple statistical-mechanics approach, we study the effect of conformation and flexibility on the quality of recognition processes. Our model relates specificity to the conformation of the participant molecules and thus suggests a possible answer: Optimal specificity is achieved when the ligand is slightly off target, that is a conformational mismatch between the ligand and its main target improves the selectivity of the process. This indicates that deformations upon binding serve as a conformational proofreading mechanism, which may be selected for via evolution. Introduction Practically all biological systems rely on the ability of bio-molecules to specifically recognize each other. Examples are antibodies targeting antigens, regulatory proteins binding DNA and enzymes catalyzing their substrates. These and other molecular recognizers must locate and preferentially interact with their specific targets among a vast variety of molecules that are often structurally similar. This task is further complicated by the inherent noise in the biochemical environment, whose magnitude is comparable with that of the non- covalent binding interactions [1-3]. -

Large-Scale Analyses of Site-Specific Evolutionary Rates Across
G C A T T A C G G C A T genes Article Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains Joseph B. Ahrens, Jordon Rahaman and Jessica Siltberg-Liberles * Department of Biological Sciences, Florida International University, Miami, FL 33199, USA; [email protected] (J.B.A.); jraha001@fiu.edu (J.R.) * Correspondence: jliberle@fiu.edu; Tel.: +1-305-348-7508 Received: 1 October 2018; Accepted: 9 November 2018; Published: 14 November 2018 Abstract: Various structural and functional constraints govern the evolution of protein sequences. As a result, the relative rates of amino acid replacement among sites within a protein can vary significantly. Previous large-scale work on Metazoan (Animal) protein sequence alignments indicated that amino acid replacement rates are partially driven by a complex interaction among three factors: intrinsic disorder propensity; secondary structure; and functional domain involvement. Here, we use sequence-based predictors to evaluate the effects of these factors on site-specific sequence evolutionary rates within four eukaryotic lineages: Metazoans; Plants; Saccharomycete Fungi; and Alveolate Protists. Our results show broad, consistent trends across all four Eukaryote groups. In all four lineages, there is a significant increase in amino acid replacement rates when comparing: (i) disordered vs. ordered sites; (ii) random coil sites vs. sites in secondary structures; and (iii) inter-domain linker sites vs. sites in functional domains. Additionally, within Metazoans, Plants, and Saccharomycetes, there is a strong confounding interaction between intrinsic disorder and secondary structure—alignment sites exhibiting both high disorder propensity and involvement in secondary structures have very low average rates of sequence evolution. -
![M.Sc. [Botany] 346 13](https://docslib.b-cdn.net/cover/3507/m-sc-botany-346-13-923507.webp)
M.Sc. [Botany] 346 13
cover page as mentioned below: below: mentioned Youas arepage instructedcover the to updateupdate to the coverinstructed pageare asYou mentioned below: Increase the font size of the Course Name. Name. 1. IncreaseCourse the theof fontsize sizefont ofthe the CourseIncrease 1. Name. use the following as a header in the Cover Page. Page. Cover 2. the usein the followingheader a as as a headerfollowing the inuse the 2. Cover Page. ALAGAPPAUNIVERSITY UNIVERSITYALAGAPPA [Accredited with ’A+’ Grade by NAAC (CGPA:3.64) in the Third Cycle Cycle Third the in (CGPA:3.64) [AccreditedNAAC by withGrade ’A+’’A+’ Gradewith by NAAC[Accredited (CGPA:3.64) in the Third Cycle and Graded as Category–I University by MHRD-UGC] MHRD-UGC] by University and Category–I Graded as as Graded Category–I and University by MHRD-UGC] M.Sc. [Botany] 003 630 – KARAIKUDIKARAIKUDI – 630 003 346 13 EDUCATION DIRECTORATEDISTANCE OF OF DISTANCEDIRECTORATE EDUCATION BIOLOGICAL TECHNIQUES IN BOTANY I - Semester BOTANY IN TECHNIQUES BIOLOGICAL M.Sc. [Botany] 346 13 cover page as mentioned below: below: mentioned Youas arepage instructedcover the to updateupdate to the coverinstructed pageare asYou mentioned below: Increase the font size of the Course Name. Name. 1. IncreaseCourse the theof fontsize sizefont ofthe the CourseIncrease 1. Name. use the following as a header in the Cover Page. Page. Cover 2. the usein the followingheader a as as a headerfollowing the inuse the 2. Cover Page. ALAGAPPAUNIVERSITY UNIVERSITYALAGAPPA [Accredited with ’A+’ Grade by NAAC (CGPA:3.64) in the Third Cycle Cycle Third the in (CGPA:3.64) [AccreditedNAAC by withGrade ’A+’’A+’ Gradewith by NAAC[Accredited (CGPA:3.64) in the Third Cycle and Graded as Category–I University by MHRD-UGC] MHRD-UGC] by University and Category–I Graded as as Graded Category–I and University by MHRD-UGC] M.Sc. -

Protein Folding and Structure Prediction
Protein Folding and Structure Prediction A Statistician's View Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Proteins Amino acids without peptide bonds. Amino acids with peptide bonds. ¡ Amino acids are the building blocks of proteins. Proteins Both figures show the same protein (the bacterial protein L). The right figure also highlights the secondary structure elements. Space Resolution limit of a light microscope Glucose Ribosome Red blood cell C−C bond Hemoglobin Bacterium 1 10 100 1000 10000 100000 1nm 1µm Distance [ A° ] Energy C−C bond Green Noncovalent bond light Glucose Thermal ATP 0.1 1 10 100 1000 Energy [ kcal/mol ] Non-Bonding Interactions Amino acids of a protein are joined by covalent bonding interactions. The polypep- tide is folded in three dimension by non-bonding interactions. These interactions, which can easily be disrupted by extreme pH, temperature, pressure, and denatu- rants, are: Electrostatic Interactions (5 kcal/mol) Hydrogen-bond Interactions (3-7 kcal/mol) Van Der Waals Interactions (1 kcal/mol) ¡ Hydrophobic Interactions ( 10 kcal/mol) The total inter-atomic force acting between two atoms is the sum of all the forces they exert on each other. Energy Profile Transition State Denatured State Native State Radius of Gyration of Denatured Proteins Do chemically denatured proteins behave as random coils? The radius of gyration Rg of a protein is defined as the root mean square dis- tance from each atom of the protein to their centroid. For an ideal (infinitely thin) random-coil chain in a solvent, the average radius 0.5 ¡ of gyration of a random coil is a simple function of its length n: Rg n For an excluded volume polymer (a polymer with non-zero thickness and non- trivial interactions between monomers) in a solvent, the average radius of gyra- 0.588 tion, we have Rg n (Flory 1953).