Response of Downy Oak (Quercus Pubescens Willd.) to Climate Change: Transcriptome Assembly, Differential Gene Analysis and Targe
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Morphometric Leaf Variation in Oaks (Quercus) of Bolu, Turkey
Ann. Bot. Fennici 40: 233–242 ISSN 0003-3847 Helsinki 29 August 2003 © Finnish Zoological and Botanical Publishing Board 2003 Morphometric leaf variation in oaks (Quercus) of Bolu, Turkey Aydın Borazan & Mehmet T. Babaç Department of Biology, Abant |zzet Baysal University, Gölköy 14280 Bolu, Turkey (e-mail: [email protected], [email protected]) Received 16 Sep. 2002, revised version received 7 Jan. 2003, accepted 10 Jan. 2003 Borazan, A. & Babaç, M. T. 2003: Morphometric leaf variation in oaks (Quercus) of Bolu, Turkey. — Ann. Bot. Fennici 40: 233–242. Genus Quercus (Fagaceae) has a problematic taxonomy because of widespread hybridization between the infrageneric taxa. The pattern of morphological leaf varia- tion was evaluated for evidence of hybridization in Bolu, Turkey, since previous stud- ies suggested that in oaks leaf morphology is a good indicator of putative hybridiza- tion. Principal components analysis was applied to data sets of leaf characters from fi ve populations to describe variation in leaf morphology. Leaf characters analyzed in this study showed high degrees of variation as a result of hybridization between four taxa (Q. pubescens, Q. virgiliana, Q. petraea and Q. robur) of subgenus Quercus while Q. cerris as a member of subgenus Cerris was clearly separated from the others. Key words: hybridization, morphological leaf variation, principal components analy- sis, Quercus Introduction in regions of mild and warm temperate climates. Fossil leaves indicate that todayʼs several major In the northern hemisphere oaks (Quercus) are oak groups are at least 40 million years old. Gen- conspicuous members of the temperate decidu- eral distribution of fossil ancestors supports the ous, broad leaved forests. -

What Is a Tree in the Mediterranean Basin Hotspot? a Critical Analysis
Médail et al. Forest Ecosystems (2019) 6:17 https://doi.org/10.1186/s40663-019-0170-6 RESEARCH Open Access What is a tree in the Mediterranean Basin hotspot? A critical analysis Frédéric Médail1* , Anne-Christine Monnet1, Daniel Pavon1, Toni Nikolic2, Panayotis Dimopoulos3, Gianluigi Bacchetta4, Juan Arroyo5, Zoltán Barina6, Marwan Cheikh Albassatneh7, Gianniantonio Domina8, Bruno Fady9, Vlado Matevski10, Stephen Mifsud11 and Agathe Leriche1 Abstract Background: Tree species represent 20% of the vascular plant species worldwide and they play a crucial role in the global functioning of the biosphere. The Mediterranean Basin is one of the 36 world biodiversity hotspots, and it is estimated that forests covered 82% of the landscape before the first human impacts, thousands of years ago. However, the spatial distribution of the Mediterranean biodiversity is still imperfectly known, and a focus on tree species constitutes a key issue for understanding forest functioning and develop conservation strategies. Methods: We provide the first comprehensive checklist of all native tree taxa (species and subspecies) present in the Mediterranean-European region (from Portugal to Cyprus). We identified some cases of woody species difficult to categorize as trees that we further called “cryptic trees”. We collected the occurrences of tree taxa by “administrative regions”, i.e. country or large island, and by biogeographical provinces. We studied the species-area relationship, and evaluated the conservation issues for threatened taxa following IUCN criteria. Results: We identified 245 tree taxa that included 210 species and 35 subspecies, belonging to 33 families and 64 genera. It included 46 endemic tree taxa (30 species and 16 subspecies), mainly distributed within a single biogeographical unit. -

From Genes to Genomes: Botanic Gardens Embracing New Tools for Conservation and Research Volume 18 • Number 1
Journal of Botanic Gardens Conservation International Volume 18 • Number 1 • February 2021 From genes to genomes: botanic gardens embracing new tools for conservation and research Volume 18 • Number 1 IN THIS ISSUE... EDITORS Suzanne Sharrock EDITORIAL: Director of Global Programmes FROM GENES TO GENOMES: BOTANIC GARDENS EMBRACING NEW TOOLS FOR CONSERVATION AND RESEARCH .... 03 Morgan Gostel Research Botanist, FEATURES Fort Worth Botanic Garden Botanical Research Institute of Texas and Director, GGI-Gardens NEWS FROM BGCI .... 06 Jean Linksy FEATURED GARDEN: THE NORTHWESTERN UNIVERSITY Magnolia Consortium Coordinator, ECOLOGICAL PARK & BOTANIC GARDENS .... 09 Atlanta Botanical Garden PLANT HUNTING TALES: GARDENS AND THEIR LESSONS: THE JOURNAL OF A BOTANY STUDENT Farahnoz Khojayori .... 13 Cover Photo: Young and aspiring scientists assist career scientists in sampling plants at the U.S. Botanic Garden for TALKING PLANTS: JONATHAN CODDINGTON, the Global Genome Initiative (U.S. Botanic Garden). DIRECTOR OF THE GLOBAL GENOME INITIATIVE .... 16 Design: Seascape www.seascapedesign.co.uk BGjournal is published by Botanic Gardens Conservation International (BGCI). It is published twice a year. Membership is open to all interested individuals, institutions and organisations that support the aims of BGCI. Further details available from: ARTICLES • Botanic Gardens Conservation International, Descanso House, 199 Kew Road, Richmond, Surrey TW9 3BW UK. Tel: +44 (0)20 8332 5953, Fax: +44 (0)20 8332 5956, E-mail: [email protected], www.bgci.org BANKING BOTANICAL BIODIVERSITY WITH THE GLOBAL GENOME • BGCI (US) Inc, The Huntington Library, BIODIVERSITY NETWORK (GGBN) Art Collections and Botanical Gardens, Ole Seberg, Gabi Dröge, Jonathan Coddington and Katharine Barker .... 19 1151 Oxford Rd, San Marino, CA 91108, USA. -

The Clinical Significance of the Organic Acids Test
The Clinical Significance of the Organic Acids Test The Organic Acids Test (OAT) provides an accurate metabolic snapshot of what is going on in the body. Besides offering the most complete and accurate evaluation of intestinal yeast and bacteria, it also provides information on important neurotransmitters, nutritional markers, glutathione status, oxalate metabolism, and much more. The test includes 76 urinary metabolite markers that can be very useful for discovering underlying causes of chronic illness. Patients and physicians report that treating yeast and bacterial abnormalities reduces fatigue, increases alertness and energy, improves sleep, normalizes bowel function, and reduces hyperactivity and abdominal pain. The OAT Assists in Evaluating: ■ Krebs Cycle Abnormalities ■ Neurotransmitter Levels ■ Nutritional Deficiencies ■ Antioxidant Deficiencies ■ Yeast and Clostridia Overgrowth ■ Fatty Acid Metabolism ■ Oxalate Levels ■ And More! The OAT Pairs Well with the Following Tests: ■ GPL-TOX: Toxic Non-Metal Chemical Profile ■ IgG Food Allergy + Candida ■ MycoTOX Profile ■ Phospholipase A2 Activity Test Learn how to better integrate the OAT into your practice, along with our other top tests by attending one of our GPL Academy Practitioner Workshops! Visit www.GPLWorkshops.com for workshop dates and locations. The following pages list the 76 metabolite markers of the Organic Acids Test. Included is the name of the metabolic marker, its clinical significance, and usual initial treatment. INTESTINAL MICROBIAL OVERGROWTH Yeast and Fungal Markers Elevated citramalic acid is produced mainly by Saccharomyces species or Propionibacteria Citramalic Acid overgrowth. High-potency, multi-strain probiotics may help rebalance GI flora. A metabolite produced by Aspergillus and possibly other fungal species in the GI tract. 5-Hydroxy-methyl- Prescription or natural antifungals, along with high-potency, multi-strain probiotics, furoic Acid may reduce overgrowth levels. -
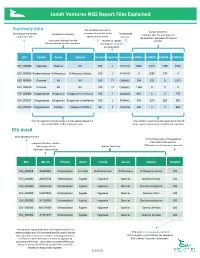
NGS Report Explained
Jonah Ventures NGS Report Files Explained Summary data The % of base pairs in the Sample identifiers Exact Sequence Variant sequence that match to the The detected Exact Sequence Variant Consensus Taxonomy The detected Numbers after the decimal point unique identifier species in the library. sequence. represent lab replicates of the same Taxonomic ranking from the Number of species sample. library matched to each sequence. matching the sequence at a given level. ESV Family Genus Species %match # Species Sequence S10001.1 S10002.1 S10003.1 S10003.2 ESV_000080 Fagaceae Quercus NA 100 7 AAAAAG… 3066 1340 5780 3462 ESV_000818 Orobanchaceae Orthocarpus Orthocarpus luteus 100 1 AAAAAG… 0 2582 249 0 ESV_000006 Poaceae NA NA 100 77 GAAAAG… 584 101 0 1039 ESV_000050 Poaceae NA NA 100 12 GAAAAG… 1328 0 0 0 ESV_000308 Polygonaceae Polygonum Polygonum humifusum 100 2 AAAAAG… 902 0 0 278 ESV_000027 Polygonaceae Eriogonum Eriogonum umbellatum 100 1 ATAAAG… 341 124 282 381 ESV_000520 Polygonaceae Fallopia Fallopia multiflora 99 1 AAAAAG… 126 0 0 994 “NA” at a taxonomic ranks means multiple species present in The number in each cell is the absolute number of the sample differ in that taxonomic rank times a given sequence was read by the sequencer. ESV detail Exact Sequence Variant The % of base pairs in the sequence that match to the species. Index identification number. that match to the species. Each sample has an Species Taxonomy 100% indicates all base pairs matched individual index number. ESV GB_ID Phylum Order Family Genus Species %match ESV_000818 -

3-Aroyl Pyroglutamic Acid Amides †
Proceeding Paper Synthesis of (2S,3S)-3-Aroyl Pyroglutamic Acid Amides † Lucia Pincekova * and Dusan Berkes * Department of Organic Chemistry, Slovak Technical University, Radlinského 9, SK-812 37 Bratislava, Slovakia * Correspondence: [email protected] (L.P.); [email protected] (D.B.) † Presented at the 24th International Electronic Conference on Synthetic Organic Chemistry, 15 November–15 December 2020; Available online: https://ecsoc-24.sciforum.net/. Abstract: A new methodology for the asymmetric synthesis of enantiomerically enriched 3-aroyl pyroglutamic acid derivatives has been developed through effective 5-exo-tet cyclization of N-chlo- roacetyl aroylalanines. The three-step sequence starts with the synthesis of N-substituted (S,S)-2- amino-4-aryl-4-oxobutanoic acids via highly diastereoselective tandem aza-Michael addition and crystallization-induced diastereomer transformation (CIDT). Their N-chloroacetylation followed by base-catalyzed cyclization and ultimate acid-catalyzed removal of chiral auxiliary without loss of stereochemical integrity furnishes the target substituted pyroglutamic acids. Finally, several series of their benzyl amides were prepared as 3-aroyl analogs of known P2X7 antagonists. Keywords: pyroglutamic acid; P2X7 receptors; aza-Michael addition; CIDT; N-debenzylation 1. Introduction Pyroglutamic acid and its derivatives are a valuable class of compounds found in various natural products and pharmaceuticals, representing either an important chiral Citation: Pincekova, L.; Berkes, D. auxiliary or building block for the asymmetric synthesis of many biologically and phar- Synthesis of (2S,3S)-3-Aroyl maceutically valuable compounds [1]. Moreover, pyroglutamic acid derivatives have re- Pyroglutamic Acid Amides. Chem. cently appeared to be efficient antagonists of specific types of purinergic receptors. Their Proc. -

Methods, Compounds, Compositions and Vehicles for Delivering 3-Amino-1-Propanesulfonic Acid
(19) TZZ _ T (11) EP 2 862 581 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 22.04.2015 Bulletin 2015/17 A61K 47/48 (2006.01) C07K 5/06 (2006.01) C07K 5/08 (2006.01) C07C 309/15 (2006.01) (2006.01) (2006.01) (21) Application number: 14200552.9 C07D 207/16 C07D 209/20 C07D 217/24 (2006.01) C07D 233/64 (2006.01) (2006.01) (2006.01) (22) Date of filing: 12.10.2007 C07D 291/02 C07D 333/24 C12P 11/00 (2006.01) A61K 38/07 (2006.01) A61K 38/08 (2006.01) A61P 25/28 (2006.01) (84) Designated Contracting States: (72) Inventor: The designation of the inventor has not AT BE BG CH CY CZ DE DK EE ES FI FR GB GR yet been filed HU IE IS IT LI LT LU LV MC MT NL PL PT RO SE SI SK TR (74) Representative: Hoffmann Eitle Patent- und Rechtsanwälte PartmbB (30) Priority: 12.10.2006 US 851039 P Arabellastraße 30 12.04.2007 US 911459 P 81925 München (DE) (62) Document number(s) of the earlier application(s) in Remarks: accordance with Art. 76 EPC: This application was filed on 30-12-2014 as a 07875176.5 / 2 089 417 divisional application to the application mentioned under INID code 62. (71) Applicant: BHI Limited Partnership Laval, QC H7V 4A7 (CA) (54) Methods, compounds, compositions and vehicles for delivering 3- amino-1-propanesulfonic acid (57) The invention relates to methods, compounds, that will yield or generate 3APS, either in vitro or in vivo. -

Quercus Robur and Quercus Petraea
Quercus robur and Quercus petraea Quercus robur and Quercus petraea in Europe: distribution, habitat, usage and threats E. Eaton, G. Caudullo, S. Oliveira, D. de Rigo these oaks can mix, compete and naturally hybridise with other Mediterranean oaks, such as Quercus pubescens and Quercus Quercus robur L., (pedunculate oak) and Quercus petraea (Matt.) Liebl., (sessile oak) are common broadleaved tree frainetto, even if at relatively low rates12. Both oaks occur at species in Europe, found from Scandinavia to the Iberian Peninsula. The two species are quite similar in appearance and higher elevations in southern regions: Q. robur is recorded to grow have a broadly overlapping range. Oak trees have cultural significance for people throughout Europe and the trees or up to 1 300 m in the Alps13, while Q. petraea is more montane leaves are frequently used in national or regional symbols. Oak trees can live for more than 1 000 years and grow to and in southern Turkey can reach over 2 000 m4, 14, 15. Due to the be 30 to 40 m in height. The wood from oaks is hard and durable and has been valued for centuries. It is favoured for substantial human interest and usage of the species over many construction and for wine and spirit barrels; historically it was a major source of ship timbers. Recently, concerns have centuries, there is widespread disturbance in their distribution, and arisen about the fate of oaks in the face of Acute Oak Decline, a little understood syndrome. the structure of their original forests is highly uncertain16. Q. -

Quercus Pubescens), Pedunculate Oak (Q
GEOCHRONOMETRIA 26 (2007), pp 39-45 DOI 10.2478/v10003-007-0005-2 Available online at versita.metapress.com and www.geochronometria.pl TREE-RING CHRONOLOGIES OF DOWNY OAK (QUERCUS PUBESCENS), PEDUNCULATE OAK (Q. ROBUR) AND SESSILE OAK (Q. PETRAEA) IN THE BIELINEK NATURE RESERVE: COMPARISON OF THE CLIMATIC DETERMINANTS OF TREE-RING WIDTH ANNA CEDRO Department of Climatology and Marine Meteorology, Institute of Marine Sciences, University of Szczecin, Wąska 13, 71-415 Szczecin Received 2 October 2006 Accepted 18 December 2006 Abstract: In 2004-2006, 50 trees of downy oak (Quercus pubescens), pedunculate oak (Q. robur) and sessile oak (Q. petraea) were sampled in the Bielinek Nature Reserve. The following chronologies were established from their tree-ring series: OMS of 212 years (1793-2004) for downy oaks well as D10 of 183 years (1817-1999) and D13 of 211 years (1789-1999) for the two indigenous oak species pedunculate and sessile oak, respectively. These chronologies were used for signature years and re- sponse function analyses. All three chronologies were highly similar, which points to identical re- sponses to the ambient meteorological conditions. The radial increment of downy oak depends pri- marily on the amount of precipitation. A high annual sum of precipitation, copious rain in spring- summer in particular, results in wide tree rings. Precipitation in form of rain and snow in winter, too, enhance tree growth by raising the groundwater level and improving the water supply in the habitat during the subsequent spring. Droughts in spring and summer, coupled with high air temperature, re- sult in narrow rings. -

Interpretive Guide
INTERPRETIVE GUIDE Contents INTRODUCTION .........................................................................1 NUTREVAL BIOMARKERS ...........................................................5 Metabolic Analysis Markers ....................................................5 Malabsorption and Dysbiosis Markers .....................................5 Cellular Energy & Mitochondrial Metabolites ..........................6 Neurotransmitter Metabolites ...............................................8 Vitamin Markers ....................................................................9 Toxin & Detoxification Markers ..............................................9 Amino Acids ..........................................................................10 Essential and Metabolic Fatty Acids .........................................13 Cardiovascular Risk ................................................................15 Oxidative Stress Markers ........................................................16 Elemental Markers ................................................................17 Toxic Elements .......................................................................18 INTERPRETATION-AT-A-GLANCE .................................................19 REFERENCES .............................................................................23 INTRODUCTION A shortage of any nutrient can lead to biochemical NutrEval profile evaluates several important biochemical disturbances that affect healthy cellular and tissue pathways to help determine nutrient -

Using Hierarchical Spatial Models to Assess the Occurrence of an Island Endemism: the Case of Salamandra Corsica Daniel Escoriza1* and Axel Hernandez2
Escoriza and Hernandez Ecological Processes (2019) 8:15 https://doi.org/10.1186/s13717-019-0169-5 RESEARCH Open Access Using hierarchical spatial models to assess the occurrence of an island endemism: the case of Salamandra corsica Daniel Escoriza1* and Axel Hernandez2 Abstract Background: Island species are vulnerable to rapid extinction, so it is important to develop accurate methods to determine their occurrence and habitat preferences. In this study, we assessed two methods for modeling the occurrence of the Corsican endemic Salamandra corsica, based on macro-ecological and fine habitat descriptors. We expected that models based on habitat descriptors would better estimate S. corsica occurrence, because its distribution could be influenced by micro-environmental gradients. The occurrence of S. corsica was modeled according to two ensembles of variables using random forests. Results: Salamandra corsica was mainly found in forested habitats, with a complex vertical structure. These habitats are associated with more stable environmental conditions. The model based on fine habitat descriptors was better able to predict occurrence, and gave no false negatives. The model based on macro-ecological variables underestimated the occurrence of the species on its ecological boundary, which is important as such locations may facilitate interpopulation connectivity. Conclusions: Implementing fine spatial resolution models requires greater investment of resources, but this is advisable for study of microendemic species, where it is important to reduce type II error (false negatives). Keywords: Amphibian, Corsica, Mediterranean islands, Microhabitat, Salamander Introduction reason, studies evaluating the niche of ectothermic verte- Macro-ecological models are popular for evaluating eco- brates associated with forests should include parameters logical and evolutionary hypotheses for vertebrates. -

Scolytus Intricatus (Ratzeburg) Coleoptera: Curculionidae European Oak Bark Beetle
Scolytus intricatus (Ratzeburg) Coleoptera: Curculionidae European Oak Bark Beetle CAPS-Approved Survey Host(s) Method Major/Primary hosts Visual Castanea sativa (Sweet chestnut), Corylus colurna, Quercus spp. (Oak), Quercus coccifera (Kermes oak), Quercus dalechampii (Dalechamp’s oak), Quercus ilex (Holly oak), Quercus petraea (Durmast oak), Quercus robur (Common oak) Other hosts Aesculus spp. (Buckeye), Aesculus hippocastanum (Horse chestnut), Alnus glutinosa (European alder), Betula spp. (Birch), Betula celtiberica (Iberian white birch), Betula pendula (Common silver birch), Betula pubescens (Downy birch), Betula verrucosa (European white birch), Carpinus spp. (Hornbeam), Carpinus betulus (European hornbeam), Castanea spp. (Chestnut), Castanea vesca (Sweet chestnut), Corylus spp. (Hazelnut), Fagus spp. (Beech), Fagus moesiaca, Fagus orientalis (Oriental beech), Fagus sylvatica (Common beech), Ostrya spp. (Hop-hornbeam), Ostrya carpinifolia (Hop-hornbeam), Parrotia persica (Persian parrotia), Populus spp. (Poplars), Populus alba (White poplar), Populus tremula (European aspen), Quercus canariensis (Algerian oak), Quercus castaneaefolia (Chestnut-leaf oak), Quercus cerris (European turkey oak), Quercus frainetto (Italian oak), Quercus hartwissiana, 1 Quercus lusitanica (Lusitanian oak), Quercus polycarpa, Quercus prinus var. tomentosa, Quercus pubescens (Downy oak), Quercus pyrenaica (Pyrenean oak), Quercus rubra (Northern red oak), Quercus virgiliana (Italian oak), Salix spp. (Willow), Sorbus spp. (Mountain Ash), Tilia cordata (Small-leaf lime), Ulmus spp. (Elms), Ulmus carpinifolia (English elm), Ulmus laevis (European white elm), Zelkova carpinifolia (Caucasian zelkova) (Davis et al., 2006; Kimoto and Duthie-Holt, 2006; CABI, 2008) Reason for Inclusion in Manual Scolytus intricatus was added to the manual in 2010. Scolytus intricatus is a target pest on the FY2011 AHP Prioritized Pest List. Pest Description Eggs: Eggs are a pearly white color and oval 1 with a length of 1 mm (< /16 in) (Haack, 2001).