Separation and Purification Methods
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Extraction of Wax from Sorghum Bran •
i EXTRACTION OF WAX FROM SORGHUM BRAN • 1 HSIEN-WEN HSU <& . ***£/ B. S., National Taiwan University, China, 1951 A THESIS » submitted in partial fulfillment of the requirements for the degree MASTER OF SCIENCE - Department of Chemical Engineering KANSAS STATE COLLEGE OF AGRICULTURE AND APPLIED SCIENCE - 1955 » k Lo i~i ii Q^cu^v^Js TABLE OF CONTENTS INTRODUCTION 1 PREVIOUS WORK 3 MATERIALS 6 GLASS SOXHLET EXTRACTION AND ANALYSIS OF WAX AND OIL IN MISCELLA 7 Procedure of Using Soxhlet Extractor 7 Separation of Wax from Oil by the Acetone Method.. 8 Summary of Results 9 PILOT PLANT EXTRACTION 15 SEPARATION OF WAX AND OIL 16 Effect of Temperature in the Acetone Method 16 Urea Complex Method of Separation. 17 Theory 17 Experimental Procedure 22 Summary of Results 26 COST ESTIMATION OF A WAX RECOVERY PLANT 29 DISCUSSION 30 CONCLUSIONS 35 ACKNOWLEDGMENTS 36 BIBLIOGRAPHY 37 APPENDIX 39 INTRODUCTION i One of the functions of the Agricultural Experiment Station is the never ending search for means of better and fuller utilization of the agri- cultural products that are of particular interest in the state of Kansas. In line with this broad objective, the extraction of economically valuable lipid materials from the sorghum bran has been studied in several theses in the Departments of Chemical Engineering and Chemistry in recent years. Generally the emphasis has b. en placed upon either the chemical and physical properties of these lipides or the equipment design and performance for the solvent ex- traction operation. A brief review of the previous work is presented in the ensuing section. -

The Separation of Three Azeotropes by Extractive Distillation by An-I Yeh A
The separation of three azeotropes by extractive distillation by An-I Yeh A thesis submitted in partial fulfillment of the requirement for the degree of Master of Science in Chemical Engineering Montana State University © Copyright by An-I Yeh (1983) Abstract: Several different kinds of extractive distillation agents were investigated to affect the separation of three binary liquid mixtures, isopropyl ether - acetone, methyl acetate - methanol, and isopropyl ether - methyl ethyl ketone. Because of the small size of the extractive distillation column, relative volatilities were assumed constant and the Fenske equation was used to calculate the relative volatilities and the number of minimum theoretical plates. Dimethyl sulfoxide was found to be a good extractive distillation agent. Extractive distillation when employing a proper agent not only negated the azeotropes of the above mixtures, but also improved the efficiency of separation. This process could reverse the relative volatility of isopropyl ether and acetone. This reversion was also found in the system of methyl acetate and methanol when nitrobenzene was the agent. However, normal distillation curves were obtained for the system of isopropyl ether and methyl ethyl ketone undergoing extractive distillation. In the system of methyl acetate and methanol, the relative volatility decreased as the agents' carbon number increased when glycols were used as the agents. In addition, the oxygen number and the locations of hydroxyl groups in the glycols used were believed to affect the values of relative volatility. An appreciable amount of agent must be maintained in the column to affect separation. When dimethyl sulfoxide was an agent for the three systems studied, the relative volatility increased as the addition rate increased. -

Development of a Process for N-Butanol Recovery from Abe
937 A publication of CHEMICAL ENGINEERING TRANSACTIONS VOL. 74, 2019 The Italian Association of Chemical Engineering Online at www.cetjournal.it Guest Editors: Sauro Pierucci, Jiří Jaromír Klemeš, Laura Piazza Copyright © 2019, AIDIC Servizi S.r.l. ISBN 978-88-95608-71-6; ISSN 2283-9216 DOI: 10.3303/CET1974157 Development of a Process for N-Butanol Recovery from Abe Wastewater Streams by Membrane Technology * Marco Stoller , Marco Bravi, Paola Russo, Roberto Bubbico, Barbara Mazzarotta Sapienza University of Rome, Dept. of Chemical Materials Environmental Engineering, Via Eudossiana 18, 00184 Rome, Italy [email protected] The aceton-butyl-ethanolic fermentation process (ABE) is a biotechnological process that leads to the production of acetone, n-butanol and ethanol (ABE compounds) from glucose sources and amides by use of certain biomasses. The process was developed initially during the middle of the last century and suffers from decline due to the greater petrochemical production of products and the lowering of the costs of the sector. Nowadays, the ABE process is regaining great interest because the fraction with the highest concentration, i.e. n-butanol, is an excellent constituent for biofuels. The ABE process has been optimized over time to obtain maximum yields of n-butanol, but the problem of separating and concentrating the butanol in the outlet stream of the ABE process persists. To allow an adequate use, often distillation by use of more columns is required. Moreover, the contained biomasses and suspended solids, in high quantity, must be eliminated, leading to overall high treatment costs. This work will report the main idea and some preliminary experimental results for the development and application of a process based on membrane technologies, to separate and concentrate the butanol from ABE process streams to sensibly reduce the difficulty to perform a final distillation. -

Distillation 65 Chem 355 Jasperse DISTILLATION
Distillation 65 Chem 355 Jasperse DISTILLATION Background Distillation is a widely used technique for purifying liquids. The basic distillation process involves heating a liquid such that liquid molecules vaporize. The vapors produced are subsequently passed through a water-cooled condenser. Upon cooling, the vapor returns to it’s liquid phase. The liquid can then be collected. The ability to separate mixtures of liquids depends on differences in volatility (the ability to vaporize). For separation to occur, the vapor that is condensed and collected must be more pure than the original liquid mix. Distillation can be used to remove a volatile solvent from a nonvolatile product; to separate a volatile product from nonvolatile impurities; or to separate two or more volatile products that have sufficiently different boiling points. Vaporization and Boiling When a liquid is placed in a closed container, some of the molecules evaporate into any unoccupied space in the container. Evaporation, which occurs at temperatures below the boiling point of a compound, involves the transition from liquid to vapor of only those molecules at the liquid surface. Evaporation continues until an equilibrium is reached between molecules entering and leaving the liquid and vapor states. The pressure exerted by these gaseous molecules on the walls of the container is the equilibrium vapor pressure. The magnitude of this vapor pressure depends on the physical characteristics of the compound and increases as temperature increases. In an open container, equilibrium is never established, the vapor can simply leave, and the liquid eventually disappears. But whether in an open or closed situation, evaporation occurs only from the surface of the liquid. -
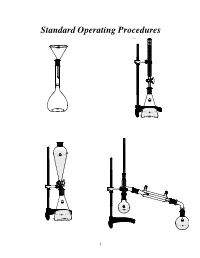
Standard Operating Procedures
Standard Operating Procedures 1 Standard Operating Procedures OVERVIEW In the following laboratory exercises you will be introduced to some of the glassware and tech- niques used by chemists to isolate components from natural or synthetic mixtures and to purify the individual compounds and characterize them by determining some of their physical proper- ties. While working collaboratively with your group members you will become acquainted with: a) Volumetric glassware b) Liquid-liquid extraction apparatus c) Distillation apparatus OBJECTIVES After finishing these sessions and reporting your results to your mentor, you should be able to: • Prepare solutions of exact concentrations • Separate liquid-liquid mixtures • Purify compounds by recrystallization • Separate mixtures by simple and fractional distillation 2 EXPERIMENT 1 Glassware Calibration, Primary and Secondary Standards, and Manual Titrations PART 1. Volumetric Glassware Calibration Volumetric glassware is used to either contain or deliver liquids at a specified temperature. Glassware manufacturers indicate this by inscribing on the volumetric ware the initials TC (to contain) or TD (to deliver) along with the calibration temperature, which is usually 20°C1. Volumetric glassware must be scrupulously clean before use. The presence of streaks or droplets is an indication of the presence of a grease film. To eliminate grease from glassware, scrub with detergent solution, rinse with tap water, and finally rinse with a small portion of distilled water. Volumetric flasks (TC) A volumetric flask has a large round bottom with only one graduation mark positioned on the long narrow neck. Graduation Mark Stopper The position of the mark facilitates the accurate and precise reading of the meniscus. If the flask is used to prepare a solution starting with a solid compound, add small amounts of sol- vent until the entire solid dissolves. -

Evaluation of Azeotropic Dehydration for the Preservation of Shrimp. James Edward Rutledge Louisiana State University and Agricultural & Mechanical College
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1969 Evaluation of Azeotropic Dehydration for the Preservation of Shrimp. James Edward Rutledge Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Rutledge, James Edward, "Evaluation of Azeotropic Dehydration for the Preservation of Shrimp." (1969). LSU Historical Dissertations and Theses. 1689. https://digitalcommons.lsu.edu/gradschool_disstheses/1689 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. This dissertation has been 70-9089 microfilmed exactly as received RUTLEDGE, James Edward, 1941- EVALUATION OF AZEOTROPIC DEHYDRATION FOR THE PRESERVATION OF SHRIMP. The Louisiana State University and Agricultural and Mechanical College, PhJD., 1969 Food Technology University Microfilms, Inc., Ann Arbor, Michigan Evaluation of Azeotropic Dehydration for the Preservation of Shrimp A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Food Science and Technology by James Edward Rutledge B.S., Texas A&M University, 1963 M.S., Texas A&M University, 1966 August, 1969 ACKNOWLEDGMENT The author wishes to express his sincere appreciation to his major professor, Dr, Fred H. Hoskins, for the guidance which he supplied not only in respect to this dissertation but also in regard to the author’s graduate career at Louisiana State University, Gratitude is also extended to Dr. -

FUGACITY It Is Simply a Measure of Molar Gibbs Energy of a Real Gas
FUGACITY It is simply a measure of molar Gibbs energy of a real gas . Modifying the simple equation for the chemical potential of an ideal gas by introducing the concept of a fugacity (f). The fugacity is an “ effective pressure” which forces the equation below to be true for real gases: θθθ f µµµ ,p( T) === µµµ (T) +++ RT ln where pθ = 1 atm pθθθ A plot of the chemical potential for an ideal and real gas is shown as a function of the pressure at constant temperature. The fugacity has the units of pressure. As the pressure approaches zero, the real gas approach the ideal gas behavior and f approaches the pressure. 1 If fugacity is an “effective pressure” i.e, the pressure that gives the right value for the chemical potential of a real gas. So, the only way we can get a value for it and hence for µµµ is from the gas pressure. Thus we must find the relation between the effective pressure f and the measured pressure p. let f = φ p φ is defined as the fugacity coefficient. φφφ is the “fudge factor” that modifies the actual measured pressure to give the true chemical potential of the real gas. By introducing φ we have just put off finding f directly. Thus, now we have to find φ. Substituting for φφφ in the above equation gives: p µ=µ+(p,T)θ (T) RT ln + RT ln φ=µ (ideal gas) + RT ln φ pθ µµµ(p,T) −−− µµµ(ideal gas ) === RT ln φφφ This equation shows that the difference in chemical potential between the real and ideal gas lies in the term RT ln φφφ.φ This is the term due to molecular interaction effects. -

United States Patent (10) Patent No.: US 9,586,922 B2 Wood Et Al
USOO9586922B2 (12) United States Patent (10) Patent No.: US 9,586,922 B2 Wood et al. (45) Date of Patent: Mar. 7, 2017 (54) METHODS FOR PURIFYING 9,388,150 B2 * 7/2016 Kim ..................... CO7D 307/48 5-(HALOMETHYL)FURFURAL 9,388,151 B2 7/2016 Browning et al. 2007/O161795 A1 7/2007 Cvak et al. 2009, 0234142 A1 9, 2009 Mascal (71) Applicant: MICROMIDAS, INC., West 2010, 0083565 A1 4, 2010 Gruter Sacramento, CA (US) 2010/0210745 A1 8/2010 McDaniel et al. 2011 O144359 A1 6, 2011 Heide et al. (72) Inventors: Alex B. Wood, Sacramento, CA (US); 2014/01 00378 A1 4/2014 Masuno et al. Shawn M. Browning, Sacramento, CA 2014/O1878O2 A1 7/2014 Mikochik et al. (US);US): Makoto N. Masuno,M. s Elk Grove, s 2015/02668432015,0203462 A1 9/20157/2015 CahanaBrowning et etal. al. CA (US); Ryan L. Smith, Sacramento, 2016/0002190 A1 1/2016 Browning et al. CA (US); John Bissell, II, Sacramento, 2016,01681 07 A1 6/2016 Masuno et al. CA (US) 2016/02O7897 A1 7, 2016 Wood et al. (73) Assignee: MICROMIDAS, INC., West FOREIGN PATENT DOCUMENTS Sacramento, CA (US) CN 101475544. A T 2009 (*) Notice: Subject to any disclaimer, the term of this N 1939: A 658 patent is extended or adjusted under 35 DE 635,783 C 9, 1936 U.S.C. 154(b) by 0 days. EP 291494 A2 11, 1988 EP 1049657 B1 3, 2003 (21) Appl. No.: 14/852,306 GB 1220851 A 1, 1971 GB 1448489 A 9, 1976 RU 2429234 C2 9, 2011 (22) Filed: Sep. -

Laboratory Rectifying Stills of Glass 2
» LABORATORY RECTIFYING STILLS OF GLASS 2 By Johannes H. Bruun 3 and Sylvester T. Schicktanz 3 ABSTRACT A complete description is given of a set of all-glass rectifying stills, suitable for distillation at pressures ranging from atmospheric down to about 50 mm. The stills are provided with efficient bubbling-cap columns containing 30 to 60 plates. Adiabatic conditions around the column are maintained by surrounding it with a jacket provided with a series of independent electrical heating units. Suitable means are provided for adjusting or maintaining the reflux ratio at the top of the column. For the purpose of conveniently obtaining an accurate value of the true boiling points of the distillates a continuous boiling-point apparatus is incor- porated in the receiving system. An efficient still of the packed-column type for distillations under pressures less than 50 mm is also described. Methods of operation and efficiency tests are given for the stills. CONTENTS Page I. Introduction 852 II. General features of the still assembly 852 III. Still pot 853 IV. Filling tube and heater for the still pot 853 V. Rectifying column 856 1. General features 856 2. Large-size column 856 3. Medium-size column 858 4. Small-size column 858 VI. Column jacket 861 VII. Reflux regulator 861 1. With variable reflux ratio 861 2. With constant reflux ratio 863 VIII. The continuous boiling-point apparatus 863 IX. Condensers and receiver 867 X. The mounting of the still 867 XI. Manufacture and transportation of the bubbling-cap still 870 XII. Operation of the bubbling-cap still 870 XIII. -

Hydro-Distillation and Steam Distillation from Aromatic Plants
Hydro-distillation and steam distillation from aromatic plants Sudeep Tandon Scientist Chemical Engineering Division CIMAP, Lucknow HISTORY Written records of herbal distillation are found as early as the first century A.D., and around 1000 A.D., the noted Arab physician and naturalist Ibn Sina also known as Avicenna described the distillation of rose oil from rose petals The ancient Arabian people began to study the chemical properties of essential oils & developed and refined the distillation process Europeans began producing essential oils in the 12th century 1 DISTILLATION ? A process in which a liquid or vapour mixture of two or more substances is separated into its component fractions of desired purity, by the application and removal of heat. In simple terms distillation of aromatic herbs implies vaporizing or liberating the oils from the trichomes / plant cell membranes of the herb in presence of high temperature and moisture and then cooling the vapour mixture to separate out the oil from water. It is the most popular widely used and cost effective method in use today for producing majority of the essential oils throughout the world Distillation is an art and not just a “Chemical" process that is reliant upon many factors for successful quality oil production. BASIC SCIENTIFIC PRINCIPLES INVOLVED IN THE PROCESS To convert any liquid into a vapour we have to apply energy in form of heat called as latent heat of vaporization A liquid always boils at the temperature at which its vapour pressure equals the atmospheric / surrounding pressure For two immiscible liquids the total vapour pressure of the mixture is always equal to the sum of their partial pressures The composition of the mixture will be determined by the concentration of the individual components into its partial pressure As known the boiling point of most essential oil components exceeds that of water and generally lies between 150 – 300oC 2 If a sample of an essential oil having a component ‘A’ having boiling point for example 190oC and the boiling point of the water is 100oC. -

By a 965% ATT'ys
July 19, 1960 A. WATZL ETAL 2,945,788 PROCESS FOR THE PURIFICATION OF DIMETHYLTEREPHTHALATE Filed Nov. 19, l956 AZEOTROPE DISTILLATE CONDENSER CONDENSED WACUUM DISTILLATE DRY DISTILLATION MXTURE N2 COLUMN MPURE - SOD WACUUMFILTER ETHYLENE DMETHYL DMETHYL GLYCOL TEREPHTHALATE TEREPHTHALATE ETHYLENE GLYCOL INVENTORS: ANTON WATZL by aERHARD 965% SIGGEL ATT'YS 2,945,788 Patented July 19, 1960 2 phthalate is immediately suitable for reesterification or Subsequent polycondensation to polyethylene terephthal 2,945,788 ate. There are gained polycondensates of high degree of PROCESS FOR THE PURIFICATION OF viscosity with K values from 50 to 57. DMETHYLTEREPHTHALATE The best mode contemplated for practicing the inven Anton Watz, Kleinwallstadt (Ufr), and Erhard Siggel, tion involves the use of ethylene glycol as the aliphatic Laudenbach (Main), Germany, assignors to Vereinigte glycol. The following is a specific illustration thereof. Glanzstoff-Fabriken A.G., Wuppertal-Elberfeld, Ger Example many Thirty grams of crude dimethylterephthalate are mixed Filed Nov.19, 1956, Ser. No. 622,778 in a flask with 270 grams of ethylene glycol and azeo tropically distilled with the introduction of dry nitrogen 4 Claims. (C. 202-42) in a column of 30 cm. at about 44 Torr (1 Torr equals 1 mm. Hg). The azeotrope goes over at about 120° C. i This invention, in general, relates to production of into a cooled condenser. The dimethylterephthalate dimethylterephthalate and more particularly to the puri 5 separated from the glycol by vacuum filtering can be fication thereof. used immediately for reesterification or for polyconden The purification of dimethylterephthalate can be car sation. This process is illustrated in the flow sheet of ried out either by distillation or by recrystallization from the accompanying drawing. -

Supercritical CO Extraction of Essential Oil from Clove
Supercritical CO2 Extraction of Essential Oil from Clove Bud: Effect of Operation Conditions on the Selective Isolation of Eugenol and Eugenyl Acetate Farshad Yazdani, Morteza Mafi, Fathollah Farhadi, Kourosh Tabar-Heidar, Kioumars Aghapoor, Farshid Mohsenzadeh, and Hossein Reza Darabi Chemistry & Chemical Engineering Research Center of Iran, P.O. Box 14335-186, Teheran, Iran Reprint requests to Dr. H. R. Darabi. E-mail: [email protected] Z. Naturforsch. 60b, 1197 – 1201 (2005); received July 14, 2005 The supercritical fluid extraction (SFE) of clove bud essential oil was studied using CO2 as sol- vent. The effect of operation conditions was analyzed in a series of experiments at temperatures between 325 and 416 K and pressures between 110 and 190 bar. The collected extracts were ana- lyzed and the relative composition of the essential oil was determined. The optimum condition was found in a temperature of 353 K and at a pressure of 190 bar, minimizing the number of extracts to two compounds (eugenol and eugenyl acetate). The extract obtained from clove bud by using su- percritical fluid extraction was compared with the essential oil obtained by steam distillation and microwave-assisted extraction by considering both quantity and quality of the product. The oil yield was higher in steam distillation and microwave oven extraction. In contrast, oil extracted by using SFE contained higher amount of eugenol and eugenyl acetate. Key words: Clove Bud, Eugenol, Extraction Introduction The high pressure is used for processes where to- Identification, extraction and preparation of clove tal extraction of a target compound is desired, since bud oil are gaining interest as its applications are most extractable compounds exhibit their maximum becoming widespread in different sectors like food, solubility in a supercritical solvent at higher pressures.