The 1257 Samalas Eruption (Lombok, Indonesia)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

435 the Date of the Minoan Santorini Eruption
THE DATE OF THE MINOAN SANTORINI ERUPTION: QUANTIFYING THE “OFFSET” Felix Höflmayer Deutsches Archäologisches Institut, Orient-Abteilung, Peter-Lenné-Strasse 32, 14195 Berlin, Germany. Email: [email protected]. ABSTRACT. Despite many recent attempts to settle the dispute concerning the absolute date of the Minoan Santorini erup- tion, there are still differences between some archaeologists and scientists on the absolute dates and the reliability of radio- carbon dating. The recent publication of over 200 new 14C dates for dynastic Egypt rules out a major flaw in the historical chronology of Egypt and proves the reliability of 14C dating in the Nile Valley. Therefore, the student of Aegean archaeology and eastern Mediterranean interconnections is still confronted with an archaeologically based conventional, or “low,” chro- nology and a 14C-backed “high” chronology. New 14C determinations from different sites of the Aegean support the high chronology for the Late Minoan (LM) IA, while recent re-evaluation of LM IB determinations are slightly higher but more or less in agreement with archaeological estimations. The present contribution reviews archaeological and scientific data for the LM IA period and argues that a reduced (~30 to 50 yr) offset between archaeological and 14C dates for the Minoan San- torini eruption may be possible, thus offering new perspectives for potential solutions for this problem. INTRODUCTION: “HIGH” AND “LOW” CHRONOLOGY AND RECENT DEVELOPMENTS Since the mid-1980s, a date for the Minoan eruption of Santorini 100 to 150 yr higher than previ- ously argued was suggested by radiocarbon dating and climatic events recorded in ice cores and tree rings from North America, Europe, and the eastern Mediterranean, placing the end of the Late Minoan (LM) IA period in the second half of the 17th century BCE (Betancourt 1987; Manning 1988, 1989a,b, 1990a,b, 1999, 2007, 2009; Michael and Betancourt 1988a,b; Friedrich et al. -

Characterization of Volcanic Deposits with Ground-Penetrating Radar
Bull Volcanol (1997) 58:515–527 Q Springer-Verlag 1997 ORIGINAL PAPER J. K. Russell 7 M. V. Stasiuk Characterization of volcanic deposits with ground-penetrating radar Received: 10 May 1996 / Accepted: 27 December 1996 Abstract Field-based studies of surficial volcanic de- Key words Ground-penetrating radar 7 Volcanic posits are commonly complicated by a combination of deposits 7 Stratigraphy 7 Dielectric constant 7 Radar poor exposure and rapid lateral variations controlled velocity 7 Pyroclastic flow 7 Airfall 7 Lava by unknown paleotopography. The potential of ground-penetrating radar (GPR) as an aid to volcano- logical studies is shown using data collected from trav- erses over four well-exposed, Recent volcanic deposits Introduction in western Canada. The deposits comprise a pumice airfall deposit (3–4 m thick), a basalt lava flow (3–6 m Studies of surficial volcanic deposits are commonly lim- thick), a pyroclastic flow deposit (15 m thick), and an ited by incomplete exposure. Estimates of the physical internally stratified pumice talus deposit (60 m thick). properties, distributions, thicknesses, and internal Results show that GPR is effective in delineating major structures of the deposits are usually derived from stud- stratigraphic contacts and hence can be used to map ies on a few well-dissected outcrops. Ground-penetrat- unexposed deposits. Different volcanic deposits also ing radar (GPR) is a portable geophysical technique exhibit different radar stratigraphic character, suggest- which can provide ancillary information on the unex- ing that deposit type may be determined from radar posed parts of deposits, thereby facilitating many volca- images. In addition, large blocks within the pyroclastic nological studies. -

And S-Wave Velocity Structures and the in Uence Of
P- and S-wave Velocity Structures and the Inuence of Volcanic Activities in the East Java Area from Seismic Tomography Syawaldin Ridha Department of Physics, Universitas Brawijaya, Indonesia Sukir Maryanto ( [email protected] ) Universitas Brawijaya https://orcid.org/0000-0002-1882-6818 Agustya A. Martha Meteorological, Climatological, and Geophysical Agency, Indonesia Vanisa Syahra Department of Physics, Universitas Brawijaya, Indonesia Muhajir Anshori Meteorological, Climatological, and Geophysical Agency, Indonesia Pepen Supendi Meteorological, Climatological, and Geophysics Agency, Indonesia Sri Widiyantoro Bandung Institute of Technology: Institut Teknologi Bandung Research Letter Keywords: seismic tomography, East Java, Vp/Vs, Vp, Vs, partial melting Posted Date: May 6th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-438689/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/24 Abstract Indonesia is one of the most interesting targets for seismic tomographic studies due to its tectonic complexity. The subduction zone was formed when the Indian oceanic plate was subducted beneath the Eurasian continental plate. This activity caused the formation of volcanoes along the Sunda Arc, including the area of East Java. In this study, we aim to identify the inuence of volcanic activities which extends from the west to the east of East Java. We used the data of 1,383 earthquakes, recorded by the 22 stations of the Indonesia Tsunami Early Warning System (InaTEWS) seismic network. We relocated the earthquakes and conducted a tomographic study using SIMULPS12. We then explored the anomalies of P- and S-wave velocities and Vp/Vs ratio. The low-velocity zone was observed in the volcanic area related to the partial melting zone or magma chamber with high Vp/Vs. -

The Minoan Eruption of Santorini, Greece
The Minoan eruption of Santorini, Greece A. BOND & R. S. J. SPARKS SUMMARY The Minoan eruption of Santorini produced logical contrasts in the mass flows which pro- the following sequence of deposits- a plinian duced them. Grain size analyses show wide pumice fall deposit, interbedded surtseyan-type ranges in the lithic contents of the different ash fall and base surge deposits, mud-flow types of deposit: ignimbrite (35--6o~), mud- deposits and ignimbrite interbedded with very flows (2o-3o ~) and the pyroclastic fall and coarse, well-sorted flood deposits. The variation base surge deposits (4-x 5 %). The ignimbrite is of thickness and grain size in the plinian deposit enriched in crystals, complemented by deple- indicates a vent x km west of Thera town. The tion in fine air-fall ash beds that interstratify base surges and surtseyan-type activity is in- with the ignimbrite. The gas velocity of the terpreted as the result of sea water entering the plinian phase is estimated as 55o m/s, the erup- magma chamber. The poorly sorted mud-flow tion column height as greater than 2o km deposits and ignimbrite are distinguished on and it is shown that only particles of 2 mm their grain size, temperature and morphological could have reached Minoan Crete. characteristics, which indicate substantial rheo- I N T H ~. L AT E B R ON Z F. A o E a paroxysmal eruption took place on Santorini Vol- cano, referred to as the Minoan eruption after the Minoan civilisation which inhabited the island at that time. The eruption produced a great volume of pumice and ash and resulted in the formation of the present day caldera which measures x 1- 5 × 8 km and probably had a catastrophic effect on the people living in the southern Aegean. -

Anatomy of a Volcanic Eruption: Case Study: Mt. St. Helens
Anatomy of a Volcanic Eruption: Case Study: Mt. St. Helens Materials Included in this Box: • Teacher Background Information • 3-D models of Mt. St. Helens (before and after eruption) • Examples of stratovolcano rock products: Tuff (pyroclastic flow), pumice, rhyolite/dacite, ash • Sandbox crater formation exercise • Laminated photos/diagrams Teacher Background There are several shapes and types of volcanoes around the world. Some volcanoes occur on the edges of tectonic plates, such as those along the ‘ring of fire’. But there are also volcanoes that occur in the middle of tectonic plates like the Yellowstone volcano and Kilauea volcano in Hawaii. When asked to draw a volcano most people will draw a steeply sided, conical mountain that has a depression (crater) at the top. This image of a 'typical' volcano is called a stratovolcano (a.k.a. composite volcano). While this is the often visualized image of a volcano, there are actually many different shapes volcanoes can be. A volcano's shape is mostly determined by the type of magma/lava that is created underneath it. Stratovolcanoes get their shape because of the thick, sticky (viscous) magma that forms at subduction zones. This magma/lava is layered between ash, pumice, and rock fragments. These layers of ash and magma will build into high elevation, steeply sided, conical shaped mountains and form a 'typical' volcano shape. Stratovolcanoes are also known for their explosive and destructive eruptions. Eruptions can cause clouds of gas, ash, dust, and rock fragments to eject into the atmosphere. These clouds of ash can become so dense and heavy that they quickly fall down the side of the volcanoes as a pyroclastic flow. -

Volcanic Eruption Impacts Student Worksheet
Volcanic Eruption Impacts Student Worksheet Explosive and Effusive Volcanoes The type of volcanic eruption is largely determined by magma composition. Flux-mediated melting at subduction zones creates a felsic magma with high levels of carbon dioxide and water. These dissolved gases explode during eruption. Effusive volcanoes have a hotter, more mafic magma with lower levels of dissolved gas, allowing them to erupt more calmly (effusive eruption). Sinabung (Indonesia) Mount Sinabung is a stratovolcano located 40 km from the Lake Toba supervolcano in North Sumatra. It lies along the Sunda Arc, where the Indo-Australian plate subducts beneath the Sunda and Burma plates. After 1200 years of dormancy, Sinabung began erupting intermittently in 2010. Major eruptions have occurred regularly since November 2013. In November and December 2015, ash plumes reached 6 – 11 km in height on multiple occasions. Pyroclastic flows and ashfall blanketed the region in January 2014 and lava flows travelled down the south flank, advancing 2.5 km by April 2014. Pyroclastic flows in February 2014 killed 17 people in a town 3 km from the vent. In June 2015, ash falls affected areas 10 – 15 km from the summit on many occasions. A lahar in May 2016, caused fatalities in a village 20 km from Sinabung. Pyroclastic flows occurred frequently throughout 2016 and 2017 Eruption of Sinabung 6 October 2016 Major eruptions occurred in 2018 and 2019. In (Y Ginsu, public domain) February 2018, an eruption destroyed a lava dome of 1.6 million cubic metres. At least 10 pyroclastic flows extended up to 4.9 km and an ash plume rose more than 16 km in altitude. -

Properties of Volcanic Ash and Pumice Concrete
Properties of volcanic ash and pumice concrete Autor(en): Hossain, Khandaker M.A. Objekttyp: Article Zeitschrift: IABSE reports = Rapports AIPC = IVBH Berichte Band (Jahr): 81 (1999) PDF erstellt am: 27.09.2021 Persistenter Link: http://doi.org/10.5169/seals-61426 Nutzungsbedingungen Die ETH-Bibliothek ist Anbieterin der digitalisierten Zeitschriften. Sie besitzt keine Urheberrechte an den Inhalten der Zeitschriften. Die Rechte liegen in der Regel bei den Herausgebern. Die auf der Plattform e-periodica veröffentlichten Dokumente stehen für nicht-kommerzielle Zwecke in Lehre und Forschung sowie für die private Nutzung frei zur Verfügung. Einzelne Dateien oder Ausdrucke aus diesem Angebot können zusammen mit diesen Nutzungsbedingungen und den korrekten Herkunftsbezeichnungen weitergegeben werden. Das Veröffentlichen von Bildern in Print- und Online-Publikationen ist nur mit vorheriger Genehmigung der Rechteinhaber erlaubt. Die systematische Speicherung von Teilen des elektronischen Angebots auf anderen Servern bedarf ebenfalls des schriftlichen Einverständnisses der Rechteinhaber. Haftungsausschluss Alle Angaben erfolgen ohne Gewähr für Vollständigkeit oder Richtigkeit. Es wird keine Haftung übernommen für Schäden durch die Verwendung von Informationen aus diesem Online-Angebot oder durch das Fehlen von Informationen. Dies gilt auch für Inhalte Dritter, die über dieses Angebot zugänglich sind. Ein Dienst der ETH-Bibliothek ETH Zürich, Rämistrasse 101, 8092 Zürich, Schweiz, www.library.ethz.ch http://www.e-periodica.ch 145 IABSE COLLOQUIUM PHUKET 1999 \ Properties of Volcanic Ash and Pumice Concrete Khandaker M. A. HOSSAIN Khandaker M. A. Hossain, born in 1963, Lecturer in Civil Engineering received BSc and MSc in civil engineering from Bangladesh University of Engineering The PNG University of Technology & Technology, Dhaka and PhD from Lae, Papua New Guinea Strathclyde University, Glasgow, UK. -

Review of Local and Global Impacts of Volcanic Eruptions and Disaster Management Practices: the Indonesian Example
geosciences Review Review of Local and Global Impacts of Volcanic Eruptions and Disaster Management Practices: The Indonesian Example Mukhamad N. Malawani 1,2, Franck Lavigne 1,3,* , Christopher Gomez 2,4 , Bachtiar W. Mutaqin 2 and Danang S. Hadmoko 2 1 Laboratoire de Géographie Physique, Université Paris 1 Panthéon-Sorbonne, UMR 8591, 92195 Meudon, France; [email protected] 2 Disaster and Risk Management Research Group, Faculty of Geography, Universitas Gadjah Mada, Yogyakarta 55281, Indonesia; [email protected] (C.G.); [email protected] (B.W.M.); [email protected] (D.S.H.) 3 Institut Universitaire de France, 75005 Paris, France 4 Laboratory of Sediment Hazards and Disaster Risk, Kobe University, Kobe City 658-0022, Japan * Correspondence: [email protected] Abstract: This paper discusses the relations between the impacts of volcanic eruptions at multiple- scales and the related-issues of disaster-risk reduction (DRR). The review is structured around local and global impacts of volcanic eruptions, which have not been widely discussed in the literature, in terms of DRR issues. We classify the impacts at local scale on four different geographical features: impacts on the drainage system, on the structural morphology, on the water bodies, and the impact Citation: Malawani, M.N.; on societies and the environment. It has been demonstrated that information on local impacts can Lavigne, F.; Gomez, C.; be integrated into four phases of the DRR, i.e., monitoring, mapping, emergency, and recovery. In Mutaqin, B.W.; Hadmoko, D.S. contrast, information on the global impacts (e.g., global disruption on climate and air traffic) only fits Review of Local and Global Impacts the first DRR phase. -
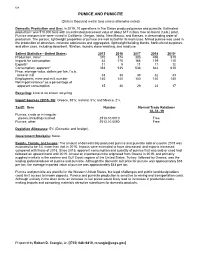
Pumice Data Sheet
128 PUMICE AND PUMICITE (Data in thousand metric tons unless otherwise noted) Domestic Production and Use: In 2019, 10 operations in five States produced pumice and pumicite. Estimated production1 was 510,000 tons with an estimated processed value of about $17 million, free on board (f.o.b.) plant. Pumice and pumicite were mined in California, Oregon, Idaho, New Mexico, and Kansas, in descending order of production. The porous, lightweight properties of pumice are well suited for its main uses. Mined pumice was used in the production of abrasives, concrete admixtures and aggregates, lightweight building blocks, horticultural purposes, and other uses, including absorbent, filtration, laundry stone washing, and road use. Salient Statistics—United States: 2015 2016 2017 2018 2019e Production, mine1 310 374 383 496 510 Imports for consumption 64 170 166 159 110 Exportse 11 9 11 11 12 Consumption, apparent2 363 535 538 644 610 Price, average value, dollars per ton, f.o.b. mine or mill 33 38 39 32 33 Employment, mine and mill, number 140 140 140 140 140 Net import reliance3 as a percentage of apparent consumption 15 30 29 23 17 Recycling: Little to no known recycling. Import Sources (2015–18): Greece, 93%; Iceland, 5%; and Mexico, 2%. Tariff: Item Number Normal Trade Relations 12–31–19 Pumice, crude or in irregular pieces, including crushed 2513.10.0010 Free. Pumice, other 2513.10.0080 Free. Depletion Allowance: 5% (Domestic and foreign). Government Stockpile: None. Events, Trends, and Issues: The amount of domestically produced pumice and pumicite sold or used in 2019 was estimated to be 3% more than that in 2018. -

Apatite Sulfur Isotope Ratios in the 1257 Samalas Eruption (Indonesia)
Goldschmidt2020 Abstract Apatite sulfur isotope ratios in the 1257 Samalas eruption (Indonesia) R. ECONOMOS1*, Y. JACKSON1, S. DING2, A. FIEGE3, C.VIDAL4, I. PRATOMO5, A. T. HERTWIG6, M.-A. LONGPRÉ2 1Southern Methodist University, Dallas, TX, USA (*[email protected]) 2CUNY Queens College, Flushing, NY, USA 3American Museum of Natural History, New York, NY, USA 4University of Cambridge, Cambridge, UK 5Geological Museum, Bandung, Indonesia 6UCLA, Los Angeles, CA, USA The 1257 eruption of Mt. Samalas in Indonesia produced sulfate anomalies in bi-polar ice cores that are ~2 times larger than those of the 1815 eruption of neighboring Tambora volcano, despite the similar magnitude of both eruptions1. The build-up of such a large volume of eruptible sulfur is likely to be related to pre-eruptive degassing and magma redox conditions. Information about these processes can be preserved in apatite crystals, which integrate sulfur as a trace element at concentrations that allow for δ34S isotope ratio characterization in-situ via Secondary Ionization Mass Spectrometry2. We investigated apatite crystals occuring as inclusions in plagioclase (Pl) crystals and as apatite microphenocrysts in contact with matrix glass, from trachydacitic pumices of both the climactic 1257 eruption and an earlier 2550 B.P. event3. 34 The lowest δ S(CDT) value observed in all four sample groups is 8.5‰, which we interpret as representative of evolved magmas entering the sub-volcanic system. Pl-hosted apatite 34 inclusions from the 1257 eruption range up to a δ S(CDT) of 11‰, while microphenocrysts reach 16‰, consistent with inclusions capturing an earlier stage of magma evolution. -

Title Characteristics of Seismicity Distribution Along the Sunda Arc
Characteristics of Seismicity Distribution along the Sunda Arc: Title Some New Observations Author(s) GHOSE, Ranajit; OIKE, Kazuo Bulletin of the Disaster Prevention Research Institute (1988), Citation 38(2): 29-48 Issue Date 1988-06 URL http://hdl.handle.net/2433/124954 Right Type Departmental Bulletin Paper Textversion publisher Kyoto University Bull. Disas. Prey. Res. Inst., Kyoto Univ., Vol. 38, Part 2, No. 332, June, 1988 29 Characteristics of Seismicity Distribution along the Sunda Arc: Some New Observations By Ranajit GHOSEand Kazuo OIKE (Manuscript received March 7, 1988) Abstract Spatio-temporal variations of earthquake activity along the Sunda arc were investigated. We prepared a strain release map for this century. Adjacent to the zones of high strain release, presence of seismically quiet zones was noted. A careful inspection of the depth distribution of the earthquakes revealed that in the eastern Sunda arc, possibly there exists a zone of scarce seismicity at an interme- diate depth. We discussed the probable implications. We also analysed the patterns of temporal distributions of earthquakes at the three different seismotectonic provinces of the Sunda arc—Sumatra, Java, and the Lesser Sunda Islands. We could clearly see that, although the causative geodynamic situations for seismicity vary significantly in space along the length of the arc, the period of increase or decrease in seismicity is largely space invariant. The locally differing levels of seismicity are superposed on the common background of long period seismicity fluctuation. Finally, clustering of seismicity at some patches along the Sunda arc was studied with respect to the altimetric gravity anomaly data. We noted some apparent conformities. -

SR 53(7) 28-29.Pdf
SHORT FEATURE NIKHILANAND PANIGRAHY Mt. Tambora Volcano with its caldera after eruption HE Sun is our default light option. But what happens when But the effect was not just limited to Indonesia. The Tit does not shine? destruction was spread far and wide. Even European countries In the year 1815, the repercussion of a very unusual event could not be spared of its evil consequences. The atmosphere in was felt worldwide for a fairly long period. The incident was the West was covered by the volcanic ash of Tambora, as a result a volcanic eruption in Mount Tambora. In Indonesia, there is of which the sun-rays could not reach the surface of the Earth. an island Sumbawa in its peninsula. This is a part of the Sunda Due to reduction of solar warmth, heavy snowfall and fatal frost islands that forms a segment of Sunda Arc. This is recognised as was found even during June to August, 1816. There was intense a string of volcanic islands. cold and the situation turned worse as famine-conditions In Mount Tambora, there exists a stratovolcano. It contains developed in European and North American countries. lava, pumice, volcanic ash, other materials and different gases. Many people from England started to rush to the lake side In many cases, a volcano looks like a cone, as the hot liquid lava of Geneva, Switzerland for warm climatic conditions. Such a emerging from it cannot fl ow to long distances away from the discouraging state of affair and unfair weather continued for vent or opening of the volcano, due to large viscosity.