Identifying Untapped Microbial Resources in the Marine Sponge Microbiome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Use of Purple Nonsulfur Photosynthetic Bacteria to Maintain Water Quality, Sources of Single Cell Protein and Bioactive Compounds for Shrimp Cultivation
The Use of Purple Nonsulfur Photosynthetic Bacteria to Maintain Water Quality, Sources of Single Cell Protein and Bioactive Compounds for Shrimp Cultivation Supaporn Chumpol A Thesis Submitted in Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Microbiology Prince of Songkla University 2017 Copyright of Prince of Songkla University i The Use of Purple Nonsulfur Photosynthetic Bacteria to Maintain Water Quality, Sources of Single Cell Protein and Bioactive Compounds for Shrimp Cultivation Supaporn Chumpol A Thesis Submitted in Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Microbiology Prince of Songkla University 2017 Copyright of Prince of Songkla University ii Thesis Title The Use of Purple Nonsulfur Photosynthetic Bacteria to Maintain Water Quality, Sources of Single Cell Protein and Bioactive Compounds for Shrimp Cultivation Author Miss Supaporn Chumpol Major Program Microbiology Major Advisor Examining Committee: ........................................................ ..........................................Chairperson (Prof. Dr. Duangporn Kantachote) (Dr. Ampaitip Sukhoom) Co-advisor ............................................Committee (Prof. Dr. Duangporn Kantachote) ........................................................ (Prof. Dr. Hiroshi Kanzaki) ............................................Committee (Prof. Dr. Hiroshi Kanzaki) ........................................................ (Assoc. Prof. Dr. Teruhiko Nitoda) ............................................Committee -

Taxonomy and Diversity of the Sponge Fauna from Walters Shoal, a Shallow Seamount in the Western Indian Ocean Region
Taxonomy and diversity of the sponge fauna from Walters Shoal, a shallow seamount in the Western Indian Ocean region By Robyn Pauline Payne A thesis submitted in partial fulfilment of the requirements for the degree of Magister Scientiae in the Department of Biodiversity and Conservation Biology, University of the Western Cape. Supervisors: Dr Toufiek Samaai Prof. Mark J. Gibbons Dr Wayne K. Florence The financial assistance of the National Research Foundation (NRF) towards this research is hereby acknowledged. Opinions expressed and conclusions arrived at, are those of the author and are not necessarily to be attributed to the NRF. December 2015 Taxonomy and diversity of the sponge fauna from Walters Shoal, a shallow seamount in the Western Indian Ocean region Robyn Pauline Payne Keywords Indian Ocean Seamount Walters Shoal Sponges Taxonomy Systematics Diversity Biogeography ii Abstract Taxonomy and diversity of the sponge fauna from Walters Shoal, a shallow seamount in the Western Indian Ocean region R. P. Payne MSc Thesis, Department of Biodiversity and Conservation Biology, University of the Western Cape. Seamounts are poorly understood ubiquitous undersea features, with less than 4% sampled for scientific purposes globally. Consequently, the fauna associated with seamounts in the Indian Ocean remains largely unknown, with less than 300 species recorded. One such feature within this region is Walters Shoal, a shallow seamount located on the South Madagascar Ridge, which is situated approximately 400 nautical miles south of Madagascar and 600 nautical miles east of South Africa. Even though it penetrates the euphotic zone (summit is 15 m below the sea surface) and is protected by the Southern Indian Ocean Deep- Sea Fishers Association, there is a paucity of biodiversity and oceanographic data. -

Lithistid’ Tetractinellid
1 Systematics of ‘lithistid’ tetractinellid 2 demosponges from the Tropical Western 3 Atlantic – implications for phylodiversity 4 and bathymetric distribution 1,2 3 4 5 Astrid Schuster , Shirley A. Pomponi , Andrzej Pisera , Paco 5 6 1,7,8 1,8 6 Cardenas´ , Michelle Kelly , Gert Worheide¨ , and Dirk Erpenbeck 1 7 Department of Earth- & Environmental Sciences, Palaeontology and Geobiology, 8 Ludwig-Maximilians-Universitat¨ M ¨unchen, Richard-Wagner Str. 10, 80333 Munich, 9 Germany 2 10 Current address: Department of Biology, NordCEE, Southern University of Denmark, 11 Campusvej 55, 5300 M Odense, Denmark 3 12 Harbor Branch Oceanographic Institute, Florida Atlantic University, 5600 U.S. 1 North, 13 Ft Pierce, FL 34946, USA 4 14 Institute of Paleobiology, Polish Academy of Sciences, ul. Twarda 51/55, 00-818 15 Warszawa, Poland 5 16 Pharmacognosy, Department of Medicinal Chemistry, Uppsala University, Husargatan 17 3, 75123 Uppsala, Sweden 6 18 National Centre for Coasts and Oceans, National Institute of Water and Atmospheric 19 Research, Private Bag 99940, Newmarket, Auckland, 1149, New Zealand 7 20 SNSB-Bayerische Staatssammlung f ¨urPalaontologie¨ und Geologie, Richard-Wagner 21 Str. 10, 80333 Munich, Germany 8 22 GeoBio-CenterLMU, Ludwig-Maximilians-Universitat¨ M ¨unchen, Richard-Wagner Str. 10, 23 80333 Munich, Germany 24 Corresponding author: 1,8 25 Dirk Erpenbeck 26 Email address: [email protected] 27 ABSTRACT PeerJ Preprints | https://doi.org/10.7287/peerj.preprints.27673v1 | CC BY 4.0 Open Access | rec: 22 Apr 2019, publ: 22 Apr 2019 28 Background Among all present demosponges, lithistids represent a polyphyletic group with 29 exceptionally well preserved fossils dating back to the Cambrian. -

A Soft Spot for Chemistry–Current Taxonomic and Evolutionary Implications of Sponge Secondary Metabolite Distribution
marine drugs Review A Soft Spot for Chemistry–Current Taxonomic and Evolutionary Implications of Sponge Secondary Metabolite Distribution Adrian Galitz 1 , Yoichi Nakao 2 , Peter J. Schupp 3,4 , Gert Wörheide 1,5,6 and Dirk Erpenbeck 1,5,* 1 Department of Earth and Environmental Sciences, Palaeontology & Geobiology, Ludwig-Maximilians-Universität München, 80333 Munich, Germany; [email protected] (A.G.); [email protected] (G.W.) 2 Graduate School of Advanced Science and Engineering, Waseda University, Shinjuku-ku, Tokyo 169-8555, Japan; [email protected] 3 Institute for Chemistry and Biology of the Marine Environment (ICBM), Carl-von-Ossietzky University Oldenburg, 26111 Wilhelmshaven, Germany; [email protected] 4 Helmholtz Institute for Functional Marine Biodiversity, University of Oldenburg (HIFMB), 26129 Oldenburg, Germany 5 GeoBio-Center, Ludwig-Maximilians-Universität München, 80333 Munich, Germany 6 SNSB-Bavarian State Collection of Palaeontology and Geology, 80333 Munich, Germany * Correspondence: [email protected] Abstract: Marine sponges are the most prolific marine sources for discovery of novel bioactive compounds. Sponge secondary metabolites are sought-after for their potential in pharmaceutical applications, and in the past, they were also used as taxonomic markers alongside the difficult and homoplasy-prone sponge morphology for species delineation (chemotaxonomy). The understanding Citation: Galitz, A.; Nakao, Y.; of phylogenetic distribution and distinctiveness of metabolites to sponge lineages is pivotal to reveal Schupp, P.J.; Wörheide, G.; pathways and evolution of compound production in sponges. This benefits the discovery rate and Erpenbeck, D. A Soft Spot for yield of bioprospecting for novel marine natural products by identifying lineages with high potential Chemistry–Current Taxonomic and Evolutionary Implications of Sponge of being new sources of valuable sponge compounds. -

Functional Equivalence and Evolutionary Convergence In
Functional equivalence and evolutionary convergence PNAS PLUS in complex communities of microbial sponge symbionts Lu Fana,b, David Reynoldsa,b, Michael Liua,b, Manuel Starkc,d, Staffan Kjelleberga,b,e, Nicole S. Websterf, and Torsten Thomasa,b,1 aSchool of Biotechnology and Biomolecular Sciences and bCentre for Marine Bio-Innovation, University of New South Wales, Sydney, New South Wales 2052, Australia; cInstitute of Molecular Life Sciences and dSwiss Institute of Bioinformatics, University of Zurich, 8057 Zurich, Switzerland; eSingapore Centre on Environmental Life Sciences Engineering, Nanyang Technological University, Republic of Singapore; and fAustralian Institute of Marine Science, Townsville, Queensland 4810, Australia Edited by W. Ford Doolittle, Dalhousie University, Halifax, NS, Canada, and approved May 21, 2012 (received for review February 24, 2012) Microorganisms often form symbiotic relationships with eukar- ties (11, 12). Symbionts also can be transmitted vertically through yotes, and the complexity of these relationships can range from reproductive cells and larvae, as has been demonstrated in those with one single dominant symbiont to associations with sponges (13, 14), insects (15), ascidians (16), bivalves (17), and hundreds of symbiont species. Microbial symbionts occupying various other animals (18). Vertical transmission generally leads equivalent niches in different eukaryotic hosts may share func- to microbial communities with limited variation in taxonomy and tional aspects, and convergent genome evolution has been function among host individuals. reported for simple symbiont systems in insects. However, for Niches with similar selections may exist in phylogenetically complex symbiont communities, it is largely unknown how prev- divergent hosts that lead comparable lifestyles or have similar alent functional equivalence is and whether equivalent functions physiological properties. -

Proposal for a Revised Classification of the Demospongiae (Porifera) Christine Morrow1 and Paco Cárdenas2,3*
Morrow and Cárdenas Frontiers in Zoology (2015) 12:7 DOI 10.1186/s12983-015-0099-8 DEBATE Open Access Proposal for a revised classification of the Demospongiae (Porifera) Christine Morrow1 and Paco Cárdenas2,3* Abstract Background: Demospongiae is the largest sponge class including 81% of all living sponges with nearly 7,000 species worldwide. Systema Porifera (2002) was the result of a large international collaboration to update the Demospongiae higher taxa classification, essentially based on morphological data. Since then, an increasing number of molecular phylogenetic studies have considerably shaken this taxonomic framework, with numerous polyphyletic groups revealed or confirmed and new clades discovered. And yet, despite a few taxonomical changes, the overall framework of the Systema Porifera classification still stands and is used as it is by the scientific community. This has led to a widening phylogeny/classification gap which creates biases and inconsistencies for the many end-users of this classification and ultimately impedes our understanding of today’s marine ecosystems and evolutionary processes. In an attempt to bridge this phylogeny/classification gap, we propose to officially revise the higher taxa Demospongiae classification. Discussion: We propose a revision of the Demospongiae higher taxa classification, essentially based on molecular data of the last ten years. We recommend the use of three subclasses: Verongimorpha, Keratosa and Heteroscleromorpha. We retain seven (Agelasida, Chondrosiida, Dendroceratida, Dictyoceratida, Haplosclerida, Poecilosclerida, Verongiida) of the 13 orders from Systema Porifera. We recommend the abandonment of five order names (Hadromerida, Halichondrida, Halisarcida, lithistids, Verticillitida) and resurrect or upgrade six order names (Axinellida, Merliida, Spongillida, Sphaerocladina, Suberitida, Tetractinellida). Finally, we create seven new orders (Bubarida, Desmacellida, Polymastiida, Scopalinida, Clionaida, Tethyida, Trachycladida). -

Giant Barrel Sponge) Population on the Southeast Florida Reef Tract Alanna D
Nova Southeastern University NSUWorks HCNSO Student Theses and Dissertations HCNSO Student Work 7-25-2019 Spatial and temporal trends in the Xestospongia muta (giant barrel sponge) population on the Southeast Florida Reef Tract Alanna D. Waldman student, [email protected] Follow this and additional works at: https://nsuworks.nova.edu/occ_stuetd Part of the Marine Biology Commons, and the Oceanography and Atmospheric Sciences and Meteorology Commons Share Feedback About This Item NSUWorks Citation Alanna D. Waldman. 2019. Spatial and temporal trends in the Xestospongia muta (giant barrel sponge) population on the Southeast Florida Reef Tract. Master's thesis. Nova Southeastern University. Retrieved from NSUWorks, . (514) https://nsuworks.nova.edu/occ_stuetd/514. This Thesis is brought to you by the HCNSO Student Work at NSUWorks. It has been accepted for inclusion in HCNSO Student Theses and Dissertations by an authorized administrator of NSUWorks. For more information, please contact [email protected]. Thesis of Alanna D. Waldman Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science M.S. Marine Biology Nova Southeastern University Halmos College of Natural Sciences and Oceanography July 2019 Approved: Thesis Committee Major Professor: David Gilliam, Ph.D. Committee Member: Jose Lopez, Ph.D. Committee Member: Charles Messing, Ph.D. This thesis is available at NSUWorks: https://nsuworks.nova.edu/occ_stuetd/514 HALMOS COLLEGE OF NATURAL SCIENCES AND OCEANOGRAPHY Spatial and temporal trends in the Xestospongia muta (giant barrel sponge) population on the Southeast Florida Reef Tract By Alanna Denbrook Waldman Submitted to the Faculty of Halmos College of Natural Sciences and Oceanography in partial fulfillment of the requirements for the degree of Master of Science with a specialty in: Marine Biology Nova Southeastern University August 2019 Table of Contents List of Figures ............................................................................................................................... -
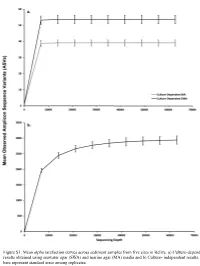
Figure S1. Mean Alpha Rarefaction Curves Across Sediment Samples from Five Sites in Belize
Figure S1. Mean alpha rarefaction curves across sediment samples from five sites in Belize. a) Culture-dependent results obtained using seawater agar (SWA) and marine agar (MA) media and b) Culture- independent results. Error bars represent standard error among replicates. a. 200 150 100 Faith’s PD Faith’s 50 0 Culturedependent MA Culturedependent SWA Cultureindependent Method b. 0.8 0.6 Pielou’s Evenness 0.4 Culturedependent MA Culturedependent SWA Cultureindependent Method Figure S2. Alpha diversity boxplots of marine sediment microbial communities from Carrie Bow Cay, Belize in culture-dependent and culture-independent samples determined using a) Faith’s Phylogenetic Diversity Index and b) Pielou’s Evenness. Culture-dependent methods include the use of marine agar medium (MA) and seawater agar medium (SWA. Data points are overlayed on the boxplot to show variation. a. 100% Proteobacteria Nitrospinae Bacteroidetes Fibrobacteres Planctomycetes Entotheonellaeota 90% Cyanobacteria Marinimicrobia (SAR406 clade) Firmicutes Deinococcus-Thermus Chloroflexi Margulisbacteria [A] Thaumarchaeota Unassigned 80% Acidobacteria Archaea UA Verrucomicrobia Acetothermia Actinobacteria Elusimicrobia 70% [A] Nanoarchaeaeota Tenericutes Kiritimatiellaeota TA06 Latescibacteria WS2 60% Spirochaetes Dependentiae Patescibacteria LCP-89 Gemmatimonadetes FCPU426 Omnitrophicaeota WPS-2 50% Fusobacteria CK-2C2-2 Bacteria UA Armatimonadetes [A] Euryarchaeota [A] Altiarchaeota Relative Percent 40% Calditrichaeota Poribacteria Lentisphaerae Cloacimonetes [A] Crenarchaeota -

Chemical and Bioactive Diversities of Marine Sponge Neopetrosia Mini
A Journal of the Bangladesh Pharmacological Society (BDPS) Bangladesh J Pharmacol 2016; 11: 433-452 Journal homepage: www.banglajol.info Abstracted/indexed in Academic Search Complete, Asia Journals Online, Bangladesh Journals Online, Biological Abstracts, BIOSIS Previews, CAB Abstracts, Current Abstracts, Directory of Open Access Journals, EMBASE/Excerpta Medica, Google Scholar, HINARI (WHO), International Pharmaceutical Abstracts, Open J-gate, Science Citation Index Expanded, SCOPUS and Social Sciences Citation Index; ISSN: 1991-0088 review - Chemical and bioactive diversities of marine sponge Neopetrosia Mini Haitham Qaralleh Department of Medical Support, Al-Balqa Applied University, Al-Karak University College, Al-Karak, Jordan. Article Info Abstract Received: 26 January 2016 The marine sponge Neopetrosia contains about 27 species that is highly Accepted: 21 March 2016 distributed in Indian Ocean, Atlantic Ocean (Caribbean Sea) and Pacific Available Online: 3 April 2016 Ocean. It has proven to be valuable to the discovery of medicinal products DOI: 10.3329/bjp.v11i2.26611 due to the presence of various types of compounds with variable bio- activities. More than 85 compounds including alkaloids, quinones, sterols and terpenoids were isolated from this genus. Moreover, the crude extracts and Cite this article: the isolated compounds revealed activities such as antimicrobial, anti-fouling, Qaralleh H. Chemical and bioactive anti-HIV, cytotoxic, anti-tumor, anti-oxidant, anti-protozoal, anti-inflamma- diversities of the marine sponge Neo- tory. Because only 9 out of 27 species of the genus Neopetrosia have been petrosia. Bangladesh J Pharmacol. chemically studied thus far, there are significant opportunities to find out new 2016; 11: 433-52. chemical constituents from this genus. -

Flavobacterial Response to Organic Pollution
Vol. 51: 31–43, 2008 AQUATIC MICROBIAL ECOLOGY Published April 24 doi: 10.3354/ame01174 Aquat Microb Ecol Flavobacterial response to organic pollution Andrew Bissett1, 2, 3,*, John P. Bowman2, Chris M. Burke1 1School of Aquaculture, Tasmanian Aquaculture and Fisheries Institute, University of Tasmania and Aquafin CRC, Launceston, Tasmania 7250, Australia 2School of Agricultural Science, University of Tasmania, Hobart, Tasmania 7001, Australia 3Present address: Max Planck Institute for Marine Microbiology, Celsiusstrasse 1, 28359 Bremen, Germany ABSTRACT: Bacteria of the Cytophaga-Flavobacterium-Bacteroides (CFB) group (phylum Bac- teroidetes), in particular members of the class Flavobacteria, are among the most prominent hetero- trophic organisms in marine pelagic systems. They have also previously been found to be important in the initial biopolymer degradation of sedimentary organic matter. The Flavobacteria community was analysed in inshore, marine sediments subject to regular inputs of highly labile organic carbon in order to understand the importance of this group in carbon degradation. We used denaturing gra- dient gel electrophoresis and real-time PCR in a statistically robust manner, over 2 consecutive years, to demonstrate that the number of Flavobacteria in the sediment increased and community composi- tion shifted with organic loading. Further community shifts occurred after cessation of organic load- ing, and population numbers also decreased. Flavobacteria appear to be important in the initial responses of the sediment microbial community to organic loading, regardless of sediment type, but flavobacterial composition was not predictable. The highly dynamic nature and large diversity (func- tional redundancy) of the Flavobacteria in these sediments may contribute to this unpredictable response. KEY WORDS: Flavobacteria . -

Implications for Dredging Management
www.nature.com/scientificreports OPEN Effects of light attenuation on the sponge holobiont- implications for dredging management Received: 08 June 2016 Mari-Carmen Pineda1,2, Brian Strehlow1,2,3, Alan Duckworth1,2, Jason Doyle1, Ross Jones1,2 & Accepted: 17 November 2016 Nicole S. Webster1,2 Published: 13 December 2016 Dredging and natural sediment resuspension events can cause high levels of turbidity, reducing the amount of light available for photosynthetic benthic biota. To determine how marine sponges respond to light attenuation, five species were experimentally exposed to a range of light treatments. Tolerance thresholds and capacity for recovery varied markedly amongst species. Whilst light attenuation had no effect on the heterotrophic speciesStylissa flabelliformis and Ianthella basta, the phototrophic species Cliona orientalis and Carteriospongia foliascens discoloured (bleached) over a 28 day exposure period to very low light (<0.8 mol photons m−2 d−1). In darkness, both species discoloured within a few days, concomitant with reduced fluorescence yields, chlorophyll concentrations and shifts in their associated microbiomes. The phototrophic species Cymbastela coralliophila was less impacted by light reduction. C. orientalis and C. coralliophila exhibited full recovery under normal light conditions, whilst C. foliascens did not recover and showed high levels of mortality. The light treatments used in the study are directly relevant to conditions that can occur in situ during dredging projects, indicating that light attenuation poses a risk to photosynthetic marine sponges. Examining benthic light levels over temporal scales would enable dredging proponents to be aware of conditions that could impact on sponge physiology. Sponges are filter-feeding organisms with important ecosystem roles including habitat provision, seawater filtra- tion, primary production and binding and/or erosion of the carbonate reef structure1. -

Compile.Xlsx
Silva OTU GS1A % PS1B % Taxonomy_Silva_132 otu0001 0 0 2 0.05 Bacteria;Acidobacteria;Acidobacteria_un;Acidobacteria_un;Acidobacteria_un;Acidobacteria_un; otu0002 0 0 1 0.02 Bacteria;Acidobacteria;Acidobacteriia;Solibacterales;Solibacteraceae_(Subgroup_3);PAUC26f; otu0003 49 0.82 5 0.12 Bacteria;Acidobacteria;Aminicenantia;Aminicenantales;Aminicenantales_fa;Aminicenantales_ge; otu0004 1 0.02 7 0.17 Bacteria;Acidobacteria;AT-s3-28;AT-s3-28_or;AT-s3-28_fa;AT-s3-28_ge; otu0005 1 0.02 0 0 Bacteria;Acidobacteria;Blastocatellia_(Subgroup_4);Blastocatellales;Blastocatellaceae;Blastocatella; otu0006 0 0 2 0.05 Bacteria;Acidobacteria;Holophagae;Subgroup_7;Subgroup_7_fa;Subgroup_7_ge; otu0007 1 0.02 0 0 Bacteria;Acidobacteria;ODP1230B23.02;ODP1230B23.02_or;ODP1230B23.02_fa;ODP1230B23.02_ge; otu0008 1 0.02 15 0.36 Bacteria;Acidobacteria;Subgroup_17;Subgroup_17_or;Subgroup_17_fa;Subgroup_17_ge; otu0009 9 0.15 41 0.99 Bacteria;Acidobacteria;Subgroup_21;Subgroup_21_or;Subgroup_21_fa;Subgroup_21_ge; otu0010 5 0.08 50 1.21 Bacteria;Acidobacteria;Subgroup_22;Subgroup_22_or;Subgroup_22_fa;Subgroup_22_ge; otu0011 2 0.03 11 0.27 Bacteria;Acidobacteria;Subgroup_26;Subgroup_26_or;Subgroup_26_fa;Subgroup_26_ge; otu0012 0 0 1 0.02 Bacteria;Acidobacteria;Subgroup_5;Subgroup_5_or;Subgroup_5_fa;Subgroup_5_ge; otu0013 1 0.02 13 0.32 Bacteria;Acidobacteria;Subgroup_6;Subgroup_6_or;Subgroup_6_fa;Subgroup_6_ge; otu0014 0 0 1 0.02 Bacteria;Acidobacteria;Subgroup_6;Subgroup_6_un;Subgroup_6_un;Subgroup_6_un; otu0015 8 0.13 30 0.73 Bacteria;Acidobacteria;Subgroup_9;Subgroup_9_or;Subgroup_9_fa;Subgroup_9_ge;