Peak Profile Analysis in X-Ray Powder Diffraction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Classical and Modern Diffraction Theory
Downloaded from http://pubs.geoscienceworld.org/books/book/chapter-pdf/3701993/frontmatter.pdf by guest on 29 September 2021 Classical and Modern Diffraction Theory Edited by Kamill Klem-Musatov Henning C. Hoeber Tijmen Jan Moser Michael A. Pelissier SEG Geophysics Reprint Series No. 29 Sergey Fomel, managing editor Evgeny Landa, volume editor Downloaded from http://pubs.geoscienceworld.org/books/book/chapter-pdf/3701993/frontmatter.pdf by guest on 29 September 2021 Society of Exploration Geophysicists 8801 S. Yale, Ste. 500 Tulsa, OK 74137-3575 U.S.A. # 2016 by Society of Exploration Geophysicists All rights reserved. This book or parts hereof may not be reproduced in any form without permission in writing from the publisher. Published 2016 Printed in the United States of America ISBN 978-1-931830-00-6 (Series) ISBN 978-1-56080-322-5 (Volume) Library of Congress Control Number: 2015951229 Downloaded from http://pubs.geoscienceworld.org/books/book/chapter-pdf/3701993/frontmatter.pdf by guest on 29 September 2021 Dedication We dedicate this volume to the memory Dr. Kamill Klem-Musatov. In reading this volume, you will find that the history of diffraction We worked with Kamill over a period of several years to compile theory was filled with many controversies and feuds as new theories this volume. This volume was virtually ready for publication when came to displace or revise previous ones. Kamill Klem-Musatov’s Kamill passed away. He is greatly missed. new theory also met opposition; he paid a great personal price in Kamill’s role in Classical and Modern Diffraction Theory goes putting forth his theory for the seismic diffraction forward problem. -

Intensity of Double-Slit Pattern • Three Or More Slits
• Intensity of double-slit pattern • Three or more slits Practice: Chapter 37, problems 13, 14, 17, 19, 21 Screen at ∞ θ d Path difference Δr = d sin θ zero intensity at d sinθ = (m ± ½ ) λ, m = 0, ± 1, ± 2, … max. intensity at d sinθ = m λ, m = 0, ± 1, ± 2, … 1 Questions: -what is the intensity of the double-slit pattern at an arbitrary position? -What about three slits? four? five? Find the intensity of the double-slit interference pattern as a function of position on the screen. Two waves: E1 = E0 sin(ωt) E2 = E0 sin(ωt + φ) where φ =2π (d sin θ )/λ , and θ gives the position. Steps: Find resultant amplitude, ER ; then intensities obey where I0 is the intensity of each individual wave. 2 Trigonometry: sin a + sin b = 2 cos [(a-b)/2] sin [(a+b)/2] E1 + E2 = E0 [sin(ωt) + sin(ωt + φ)] = 2 E0 cos (φ / 2) cos (ωt + φ / 2)] Resultant amplitude ER = 2 E0 cos (φ / 2) Resultant intensity, (a function of position θ on the screen) I position - fringes are wide, with fuzzy edges - equally spaced (in sin θ) - equal brightness (but we have ignored diffraction) 3 With only one slit open, the intensity in the centre of the screen is 100 W/m 2. With both (identical) slits open together, the intensity at locations of constructive interference will be a) zero b) 100 W/m2 c) 200 W/m2 d) 400 W/m2 How does this compare with shining two laser pointers on the same spot? θ θ sin d = d Δr1 d θ sin d = 2d θ Δr2 sin = 3d Δr3 Total, 4 The total field is where φ = 2π (d sinθ )/λ •When d sinθ = 0, λ , 2λ, .. -

Light and Matter Diffraction from the Unified Viewpoint of Feynman's
European J of Physics Education Volume 8 Issue 2 1309-7202 Arlego & Fanaro Light and Matter Diffraction from the Unified Viewpoint of Feynman’s Sum of All Paths Marcelo Arlego* Maria de los Angeles Fanaro** Universidad Nacional del Centro de la Provincia de Buenos Aires CONICET, Argentine *[email protected] **[email protected] (Received: 05.04.2018, Accepted: 22.05.2017) Abstract In this work, we present a pedagogical strategy to describe the diffraction phenomenon based on a didactic adaptation of the Feynman’s path integrals method, which uses only high school mathematics. The advantage of our approach is that it allows to describe the diffraction in a fully quantum context, where superposition and probabilistic aspects emerge naturally. Our method is based on a time-independent formulation, which allows modelling the phenomenon in geometric terms and trajectories in real space, which is an advantage from the didactic point of view. A distinctive aspect of our work is the description of the series of transformations and didactic transpositions of the fundamental equations that give rise to a common quantum framework for light and matter. This is something that is usually masked by the common use, and that to our knowledge has not been emphasized enough in a unified way. Finally, the role of the superposition of non-classical paths and their didactic potential are briefly mentioned. Keywords: quantum mechanics, light and matter diffraction, Feynman’s Sum of all Paths, high education INTRODUCTION This work promotes the teaching of quantum mechanics at the basic level of secondary school, where the students have not the necessary mathematics to deal with canonical models that uses Schrodinger equation. -

The Path Integral Approach to Quantum Mechanics Lecture Notes for Quantum Mechanics IV
The Path Integral approach to Quantum Mechanics Lecture Notes for Quantum Mechanics IV Riccardo Rattazzi May 25, 2009 2 Contents 1 The path integral formalism 5 1.1 Introducingthepathintegrals. 5 1.1.1 Thedoubleslitexperiment . 5 1.1.2 An intuitive approach to the path integral formalism . .. 6 1.1.3 Thepathintegralformulation. 8 1.1.4 From the Schr¨oedinger approach to the path integral . .. 12 1.2 Thepropertiesofthepathintegrals . 14 1.2.1 Pathintegralsandstateevolution . 14 1.2.2 The path integral computation for a free particle . 17 1.3 Pathintegralsasdeterminants . 19 1.3.1 Gaussianintegrals . 19 1.3.2 GaussianPathIntegrals . 20 1.3.3 O(ℏ) corrections to Gaussian approximation . 22 1.3.4 Quadratic lagrangians and the harmonic oscillator . .. 23 1.4 Operatormatrixelements . 27 1.4.1 Thetime-orderedproductofoperators . 27 2 Functional and Euclidean methods 31 2.1 Functionalmethod .......................... 31 2.2 EuclideanPathIntegral . 32 2.2.1 Statisticalmechanics. 34 2.3 Perturbationtheory . .. .. .. .. .. .. .. .. .. .. 35 2.3.1 Euclidean n-pointcorrelators . 35 2.3.2 Thermal n-pointcorrelators. 36 2.3.3 Euclidean correlators by functional derivatives . ... 38 0 0 2.3.4 Computing KE[J] and Z [J] ................ 39 2.3.5 Free n-pointcorrelators . 41 2.3.6 The anharmonic oscillator and Feynman diagrams . 43 3 The semiclassical approximation 49 3.1 Thesemiclassicalpropagator . 50 3.1.1 VanVleck-Pauli-Morette formula . 54 3.1.2 MathematicalAppendix1 . 56 3.1.3 MathematicalAppendix2 . 56 3.2 Thefixedenergypropagator. 57 3.2.1 General properties of the fixed energy propagator . 57 3.2.2 Semiclassical computation of K(E)............. 61 3.2.3 Two applications: reflection and tunneling through a barrier 64 3 4 CONTENTS 3.2.4 On the phase of the prefactor of K(xf ,tf ; xi,ti) .... -
Simple Cubic Lattice
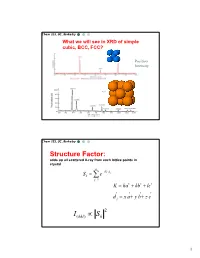
Chem 253, UC, Berkeley What we will see in XRD of simple cubic, BCC, FCC? Position Intensity Chem 253, UC, Berkeley Structure Factor: adds up all scattered X-ray from each lattice points in crystal n iKd j Sk e j1 K ha kb lc d j x a y b z c 2 I(hkl) Sk 1 Chem 253, UC, Berkeley X-ray scattered from each primitive cell interfere constructively when: eiKR 1 2d sin n For n-atom basis: sum up the X-ray scattered from the whole basis Chem 253, UC, Berkeley ' k d k d di R j ' K k k Phase difference: K (di d j ) The amplitude of the two rays differ: eiK(di d j ) 2 Chem 253, UC, Berkeley The amplitude of the rays scattered at d1, d2, d3…. are in the ratios : eiKd j The net ray scattered by the entire cell: n iKd j Sk e j1 2 I(hkl) Sk Chem 253, UC, Berkeley For simple cubic: (0,0,0) iK0 Sk e 1 3 Chem 253, UC, Berkeley For BCC: (0,0,0), (1/2, ½, ½)…. Two point basis 1 2 iK ( x y z ) iKd j iK0 2 Sk e e e j1 1 ei (hk l) 1 (1)hkl S=2, when h+k+l even S=0, when h+k+l odd, systematical absence Chem 253, UC, Berkeley For BCC: (0,0,0), (1/2, ½, ½)…. Two point basis S=2, when h+k+l even S=0, when h+k+l odd, systematical absence (100): destructive (200): constructive 4 Chem 253, UC, Berkeley Observable diffraction peaks h2 k 2 l 2 Ratio SC: 1,2,3,4,5,6,8,9,10,11,12. -

Diffraction-Free Beams in Fractional Schrödinger Equation Yiqi Zhang1, Hua Zhong1, Milivoj R
www.nature.com/scientificreports OPEN Diffraction-free beams in fractional Schrödinger equation Yiqi Zhang1, Hua Zhong1, Milivoj R. Belić2, Noor Ahmed1, Yanpeng Zhang1 & Min Xiao3,4 We investigate the propagation of one-dimensional and two-dimensional (1D, 2D) Gaussian beams in Received: 14 December 2015 the fractional Schrödinger equation (FSE) without a potential, analytically and numerically. Without Accepted: 11 March 2016 chirp, a 1D Gaussian beam splits into two nondiffracting Gaussian beams during propagation, while a 2D Published: 21 April 2016 Gaussian beam undergoes conical diffraction. When a Gaussian beam carries linear chirp, the 1D beam deflects along the trajectoriesz = ±2(x − x0), which are independent of the chirp. In the case of 2D Gaussian beam, the propagation is also deflected, but the trajectories align along the diffraction cone z =+2 xy22 and the direction is determined by the chirp. Both 1D and 2D Gaussian beams are diffractionless and display uniform propagation. The nondiffracting property discovered in this model applies to other beams as well. Based on the nondiffracting and splitting properties, we introduce the Talbot effect of diffractionless beams in FSE. Fractional effects, such as fractional quantum Hall effect1, fractional Talbot effect2, fractional Josephson effect3 and other effects coming from the fractional Schrödinger equation (FSE)4, introduce seminal phenomena in and open new areas of physics. FSE, developed by Laskin5–7, is a generalization of the Schrödinger equation (SE) that contains fractional Laplacian instead of the usual one, which describes the behavior of particles with fractional spin8. This generalization produces nonlocal features in the wave function. In the last decade, research on FSE was very intensive9–17. -

The Doubling of the Degrees of Freedom in Quantum Dissipative Systems, and the Semantic Information Notion and Measure in Biosemiotics †
Proceedings The Doubling of the Degrees of Freedom in Quantum Dissipative Systems, and the Semantic † Information Notion and Measure in Biosemiotics Gianfranco Basti 1,*, Antonio Capolupo 2,3 and GiuseppeVitiello 2 1 Faculty of Philosophy, Pontifical Lateran University, 00120 Vatican City, Italy 2 Department of Physics “E:R. Caianiello”, University of Salerno, Via Giovanni Paolo II, 132, 84084 Fisciano (SA), Italy; [email protected] (A.C.); [email protected] (G.V.) 3 Istituto Nazionale di Fisica Nucleare (INFN), Gruppo Collegato di Salerno, Via Giovanni Paolo II, 132, 84084 Fisciano (SA), Italy * Correspondence: [email protected]; Tel.: +39-339-576-0314 † Conference Theoretical Information Studies (TIS), Berkeley, CA, USA, 2–6 June 2019. Published: 11 May 2020 Keywords: semantic information; non-equilibrium thermodynamics; quantum field theory 1. The Semantic Information Issue and Non-Equilibrium Thermodynamics In the recent history of the effort for defining a suitable notion and measure of semantic information, starting points are: the “syntactic” notion and measure of information H of Claude Shannon in the mathematical theory of communications (MTC) [1]; and the “semantic” notion and measure of information content or informativeness of Rudolph Carnap and Yehoshua Bar-Hillel [2], based on Carnap’s theory of intensional modal logic [3]. The relationship between H and the notion and measure of entropy S in Gibbs’ statistical mechanics (SM) and then in Boltzmann’s thermodynamics is a classical one, because they share the same mathematical form. We emphasize that Shannon’s information measure is the information associated to an event in quantum mechanics (QM) [4]. Indeed, in the classical limit, i.e., whenever the classical notion of probability applies, S is equivalent to the QM definition of entropy given by John Von Neumann. -

Chapter 14 Interference and Diffraction
Chapter 14 Interference and Diffraction 14.1 Superposition of Waves.................................................................................... 14-2 14.2 Young’s Double-Slit Experiment ..................................................................... 14-4 Example 14.1: Double-Slit Experiment................................................................ 14-7 14.3 Intensity Distribution ........................................................................................ 14-8 Example 14.2: Intensity of Three-Slit Interference ............................................ 14-11 14.4 Diffraction....................................................................................................... 14-13 14.5 Single-Slit Diffraction..................................................................................... 14-13 Example 14.3: Single-Slit Diffraction ................................................................ 14-15 14.6 Intensity of Single-Slit Diffraction ................................................................. 14-16 14.7 Intensity of Double-Slit Diffraction Patterns.................................................. 14-19 14.8 Diffraction Grating ......................................................................................... 14-20 14.9 Summary......................................................................................................... 14-22 14.10 Appendix: Computing the Total Electric Field............................................. 14-23 14.11 Solved Problems .......................................................................................... -

Optical Solitons and Vortices in Fractional Media: a Mini-Review of Recent Results
hv photonics Review Optical Solitons and Vortices in Fractional Media: A Mini-Review of Recent Results Boris A. Malomed 1,2 1 Department of Physical Electronics, School of Electrical Engineering, Faculty of Engineering, Center for Light-Matter Interaction, Tel Aviv University, Tel Aviv P.O. Box 39040, Israel; [email protected] 2 Instituto de Alta Investigación, Universidad de Tarapacá, Casilla 7D, Arica 1000000, Chile Abstract: The article produces a brief review of some recent results which predict stable propagation of solitons and solitary vortices in models based on the nonlinear Schrödinger equation (NLSE) including fractional one-dimensional or two-dimensional diffraction and cubic or cubic-quintic nonlinear terms, as well as linear potentials. The fractional diffraction is represented by fractional- order spatial derivatives of the Riesz type, defined in terms of the direct and inverse Fourier transform. In this form, it can be realized by spatial-domain light propagation in optical setups with a specially devised combination of mirrors, lenses, and phase masks. The results presented in the article were chiefly obtained in a numerical form. Some analytical findings are included too, in particular, for fast moving solitons and the results produced by the variational approximation. Moreover, dissipative solitons are briefly considered, which are governed by the fractional complex Ginzburg–Landau equation. Keywords: fractional diffraction; nonlinear Schrödinger equation; soliton stability; collapse; symmetry breaking; complex Ginzburg–Landau equation; vortex necklaces dissipative solitons Citation: Malomed, B.A. Optical 1. Introduction and the Basic Models Solitons and Vortices in Fractional Nonlinear Schrödinger equations (NLSEs) give rise to soliton families in a great num- Media: A Mini-Review of Recent ber of realizations [1–8] , many of which originate in optics. -

Diffraction of Collinear Correlated Photon Pairs by an Ultrasonic Wave
Diffraction of collinear correlated photon pairs by an ultrasonic wave Piotr Kwiek University of Gdansk, Institute of Experimental Physics, Wita Stwosza 57, 80-952 Gdańsk, Poland e-mail address: [email protected] The phenomenon of collinear correlated photon pairs diffraction by an ultrasonic wave is investigated for Bragg incidence. A BBO crystal was used for producing collinear correlated photon pairs via type-I spontaneous parametric down-conversion (SPDC I-type). It is shown experimentally that the Bragg angle for photon pairs diffraction is identical to the one corresponding to single photons diffraction. The numbers of single photons and photon pairs counts in discrete diffraction orders were measured as functions of the Raman-Nath parameter. Similarly, the number of coincidence photon counts in separate diffraction orders was also investigated. What is more, simple analytical formulas are derived which perfectly describe experimentally obtained data. OCIS codes: (050.1940) Diffraction; (230.1040) Acousto-optical devices; (230.4110) Modulators; (270.4180) Multiphoton processes. 1. Introduction Acousto-optic devices are commonly used to modify properties of optical beams such as amplitude or intensity, phase, frequency, polarization and direction of propagation [1, 2]. Depending on which property of light is manipulated, one can name intensity modulators, phase modulators, frequency modulators, polarization modulators and spatial light modulators. Acousto- optic devices were also employed in experimental investigations devoted to photon pairs or entangled photon pairs [3-8]. Here, in most cases, acousto-optic modulators altered, at a time, parameters of just one of the photons of a given pair. Only the works of the group of Zeilinger [7] and Kwiek [8] considered simultaneous photon pairs interaction with an ultrasonic wave. -

Physics 116 Diffraction and Resolution
Physics 116 Session 25 Diffraction and resolution Nov 10, 2011 R. J. Wilkes Email: [email protected] Announcements • Posted exam score = (6 pts x number correct) + 10 pts • Scores for all 3 midterm exams will be normalized to a common average to minimize differences • Class average final grade will be 2.9 • Only your 2 best exam scores are used • Of course, if you get a perfect score on everything (exams, homeworks, quizzes), you get a 4.0, regardless of which exam was dropped! • Don’t forget: UW is closed tomorrow – no class!! Lecture Schedule (up to exam 3) Today 3 Diffraction • Everyday experience: light “gets around corners” – Shadows are not usually sharp-edged – Analogy: you can hear sound waves around the corner of a building, even if source of sound is not in your line of sight • Apply Huygens’ Principle to a single narrow slit – Picture tells us two things: 1. Spherical wavelets - some light will SLIT spherical wavelets be seen at large angles to axis MASK 2. Light from different parts of slit area will interfere plane wave So we expect to see fringes on a distant screen, including some at large angles: This kind of interference is called DIFFRACTION We see diffraction effects near any obstacle, IF we look closely enough (on a scale comparable to light wavelengths) Diffraction effects • Also see diffraction around knife-edge, needle point, etc – Shadow of knife or needle is sharp-edged only if you don’t look too closely (and use coherent or at least “monochromatic” light) –On a microscopic scale you see diffraction fringe -

Chapter 5: Interference and Diffraction
Physics 341 Experiment 5 Page 5-1 Chapter 5: Interference and Diffraction 5.1 Introduction Interference and diffraction are common phenomena intrinsic to wave propagation. Interference refers to the effects caused by the coherent addition of wave amplitudes that travel different paths. If such waves are in phase, the light intensity is enhanced; conversely if they are out of phase, the light is attenuated. Diffraction is the result of wave propagation that spreads a beam of light from a straight linear path. The origin of the two phenomena is, in any case, exactly the same. The following experiments are arranged in order of increasing complexity. This may blur the distinction in your mind between the two phenomena of interference and diffraction. That’s not entirely a bad idea, as they are so closely related. Figure 5.1 “Snapshot” of a wave Figure 5.2 The amplitude of two waves, 180o out of phase. For most of the following exercises, you will observe diffraction and interference patterns generated by a He-Ne laser beam incident on a variety of targets. These come in two forms: 2” x 2” squares such as 35-mm mounted film transparencies and rotating plastic disks imprinted with a number of selectable patterns. These are listed with dimensions in Table 5.1 and Table 5.2 below. The laser operating wavelength is 632.8 nm. With these numbers, you can compare experimental observations with theoretical estimates. A list of targets is given in Table 5.1 Note that most of the photographic targets are available as complementary pairs of positive and negative, i.e., where the positive target is transparent, the negative one is opaque and vice-versa.