An Inducible Nitrilase from a Thermophilic Bacillus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WO 2007/084545 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (43) International Publication Date (10) International Publication Number 26 July 2007 (26.07.2007) PCT WO 2007/084545 Al (51) International Patent Classification: (74) Agent: ZERULL, Susan, Moeller; The Dow Chemical C07C 227/32 (2006.01) C12P 41/00 (2006.01) Company, Intellectual Property Section, P.O. Box 1967, C07D 317/30 (2006.01) Midland, MI 48674-1967 (US). (21) International Application Number: (81) Designated States (unless otherwise indicated, for every PCT/US2007/001207 kind of national protection available): AE, AG, AL, AM, AT,AU, AZ, BA, BB, BG, BR, BW, BY, BZ, CA, CH, CN, (22) International Filing Date: 17 January 2007 (17.01.2007) CO, CR, CU, CZ, DE, DK, DM, DZ, EC, EE, EG, ES, FI, (25) Filing Language: English GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, LR, LS, (26) Publication Language: English LT, LU, LV,LY,MA, MD, MG, MK, MN, MW, MX, MY, (30) Priority Data: MZ, NA, NG, NI, NO, NZ, OM, PG, PH, PL, PT, RO, RS, 11/333,937 18 January 2006 (18.01.2006) US RU, SC, SD, SE, SG, SK, SL, SM, SV, SY, TJ, TM, TN, (71) Applicant (for all designated States except US): DOW TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW GLOBAL TECHNOLOGIES INC. [US/US]; Washing (84) Designated States (unless otherwise indicated, for every ton Street, 1790 Building, Midland, MI 48674 (US). -

The Genomes of Polyextremophilic Cyanidiales Contain 1% 2 Horizontally Transferred Genes with Diverse Adaptive Functions 3 4 Alessandro W
bioRxiv preprint doi: https://doi.org/10.1101/526111; this version posted January 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 The genomes of polyextremophilic Cyanidiales contain 1% 2 horizontally transferred genes with diverse adaptive functions 3 4 Alessandro W. Rossoni1#, Dana C. Price2, Mark Seger3, Dagmar Lyska1, Peter Lammers3, 5 Debashish Bhattacharya4 & Andreas P.M. Weber1* 6 7 1Institute of Plant Biochemistry, Cluster of Excellence on Plant Sciences (CEPLAS), Heinrich 8 Heine University, Universitätsstraße 1, 40225 Düsseldorf, Germany 9 2Department of Plant Biology, Rutgers University, New Brunswick, NJ 08901, USA 10 3Arizona Center for Algae Technology and Innovation, Arizona State University, Mesa, AZ 11 85212, USA 12 4Department of Biochemistry and Microbiology, Rutgers University, New Brunswick, NJ 13 08901, USA 14 15 *Corresponding author: Prof. Dr. Andreas P.M. Weber, 16 e-mail: [email protected] 17 bioRxiv preprint doi: https://doi.org/10.1101/526111; this version posted January 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 18 Abstract 19 The role and extent of horizontal gene transfer (HGT) in eukaryotes are hotly disputed topics 20 that impact our understanding regarding the origin of metabolic processes and the role of 21 organelles in cellular evolution. -
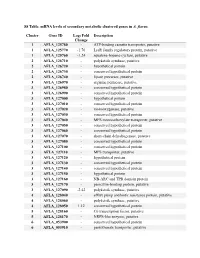
S8 Table. Mrna Levels of Secondary Metabolic Clustered Genes in A
S8 Table. mRNA levels of secondary metabolic clustered genes in A. flavus. Cluster Gene ID Log2 Fold Description Change 1 AFLA_125780 - ATP-binding cassette transporter, putative 1 AFLA_125770 -1.76 LysR family regulatory protein, putative 1 AFLA_125760 -1.24 squalene-hopene-cyclase, putative 2 AFLA_126710 - polyketide synthase, putative 2 AFLA_126720 - hypothetical protein 2 AFLA_126730 - conserved hypothetical protein 2 AFLA_126740 - lipase precursor, putative 3 AFLA_126970 - arginine permease, putative 3 AFLA_126980 - conserved hypothetical protein 3 AFLA_126990 - conserved hypothetical protein 3 AFLA_127000 - hypothetical protein 3 AFLA_127010 - conserved hypothetical protein 3 AFLA_127020 - monooxygenase, putative 3 AFLA_127030 - conserved hypothetical protein 3 AFLA_127040 - MFS monocarboxylate transporter, putative 3 AFLA_127050 - conserved hypothetical protein 3 AFLA_127060 - conserved hypothetical protein 3 AFLA_127070 - short-chain dehydrogenase, putative 3 AFLA_127080 - conserved hypothetical protein 3 AFLA_127100 - conserved hypothetical protein 3 AFLA_127110 - MFS transporter, putative 3 AFLA_127120 - hypothetical protein 3 AFLA_127130 - conserved hypothetical protein 3 AFLA_127140 - conserved hypothetical protein 3 AFLA_127150 - hypothetical protein 3 AFLA_127160 - NB-ARC and TPR domain protein 3 AFLA_127170 - penicillin-binding protein, putative 3 AFLA_127090 -2.42 polyketide synthase, putative 4 AFLA_128040 - efflux pump antibiotic resistance protein, putative 4 AFLA_128060 - polyketide synthase, putative 4 AFLA_128050 -

Comparative Genomics of Bradyrhizobium Japonicum CPAC
Siqueira et al. BMC Genomics 2014, 15:420 http://www.biomedcentral.com/1471-2164/15/420 RESEARCH ARTICLE Open Access Comparative genomics of Bradyrhizobium japonicum CPAC 15 and Bradyrhizobium diazoefficiens CPAC 7: elite model strains for understanding symbiotic performance with soybean Arthur Fernandes Siqueira1,2†, Ernesto Ormeño-Orrillo3†,RangelCelsoSouza4,ElisetePainsRodrigues5, Luiz Gonzaga Paula Almeida4, Fernando Gomes Barcellos5, Jesiane Stefânia Silva Batista6, Andre Shigueyoshi Nakatani2, Esperanza Martínez-Romero3, Ana Tereza Ribeiro Vasconcelos4 and Mariangela Hungria1,2* Abstract Background: The soybean-Bradyrhizobium symbiosis can be highly efficient in fixing nitrogen, but few genomic sequences of elite inoculant strains are available. Here we contribute with information on the genomes of two commercial strains that are broadly applied to soybean crops in the tropics. B. japonicum CPAC 15 (=SEMIA 5079) is outstanding in its saprophytic capacity and competitiveness, whereas B. diazoefficiens CPAC 7 (=SEMIA 5080) is known for its high efficiency in fixing nitrogen. Both are well adapted to tropical soils. The genomes of CPAC 15 and CPAC 7 were compared to each other and also to those of B. japonicum USDA 6T and B. diazoefficiens USDA 110T. Results: Differences in genome size were found between species, with B. japonicum having larger genomes than B. diazoefficiens. Although most of the four genomes were syntenic, genome rearrangements within and between species were observed, including events in the symbiosis island. In addition to the symbiotic region, several genomic islands were identified. Altogether, these features must confer high genomic plasticity that might explain adaptation and differences in symbiotic performance. It was not possible to attribute known functions to half of the predicted genes. -

1 Metabolic Dysfunction Is Restricted to the Sciatic Nerve in Experimental
Page 1 of 255 Diabetes Metabolic dysfunction is restricted to the sciatic nerve in experimental diabetic neuropathy Oliver J. Freeman1,2, Richard D. Unwin2,3, Andrew W. Dowsey2,3, Paul Begley2,3, Sumia Ali1, Katherine A. Hollywood2,3, Nitin Rustogi2,3, Rasmus S. Petersen1, Warwick B. Dunn2,3†, Garth J.S. Cooper2,3,4,5* & Natalie J. Gardiner1* 1 Faculty of Life Sciences, University of Manchester, UK 2 Centre for Advanced Discovery and Experimental Therapeutics (CADET), Central Manchester University Hospitals NHS Foundation Trust, Manchester Academic Health Sciences Centre, Manchester, UK 3 Centre for Endocrinology and Diabetes, Institute of Human Development, Faculty of Medical and Human Sciences, University of Manchester, UK 4 School of Biological Sciences, University of Auckland, New Zealand 5 Department of Pharmacology, Medical Sciences Division, University of Oxford, UK † Present address: School of Biosciences, University of Birmingham, UK *Joint corresponding authors: Natalie J. Gardiner and Garth J.S. Cooper Email: [email protected]; [email protected] Address: University of Manchester, AV Hill Building, Oxford Road, Manchester, M13 9PT, United Kingdom Telephone: +44 161 275 5768; +44 161 701 0240 Word count: 4,490 Number of tables: 1, Number of figures: 6 Running title: Metabolic dysfunction in diabetic neuropathy 1 Diabetes Publish Ahead of Print, published online October 15, 2015 Diabetes Page 2 of 255 Abstract High glucose levels in the peripheral nervous system (PNS) have been implicated in the pathogenesis of diabetic neuropathy (DN). However our understanding of the molecular mechanisms which cause the marked distal pathology is incomplete. Here we performed a comprehensive, system-wide analysis of the PNS of a rodent model of DN. -

Discovery of an Alternate Metabolic Pathway for Urea Synthesis in Adult Aedes Aegypti Mosquitoes
Discovery of an alternate metabolic pathway for urea synthesis in adult Aedes aegypti mosquitoes Patricia Y. Scaraffia*†‡, Guanhong Tan§, Jun Isoe*†, Vicki H. Wysocki*§, Michael A. Wells*†, and Roger L. Miesfeld*† Departments of §Chemistry and *Biochemistry and Molecular Biophysics and †Center for Insect Science, University of Arizona, Tucson, AZ 85721-0088 Edited by Anthony A. James, University of California, Irvine, CA, and approved December 4, 2007 (received for review August 27, 2007) We demonstrate the presence of an alternate metabolic pathway We previously reported that mosquitoes dispose of toxic for urea synthesis in Aedes aegypti mosquitoes that converts uric ammonia through glutamine (Gln) and proline (Pro) synthesis, acid to urea via an amphibian-like uricolytic pathway. For these along with excretion of ammonia, uric acid, and urea (20). By studies, female mosquitoes were fed a sucrose solution containing using labeled isotopes and mass spectrometry techniques (21), 15 15 15 15 15 NH4Cl, [5- N]-glutamine, [ N]-proline, allantoin, or allantoic we have recently determined how the N from NH4Cl is acid. At 24 h after feeding, the feces were collected and analyzed incorporated into the amide side chain of Gln, and then into Pro, in a mass spectrometer. Specific enzyme inhibitors confirmed that in Ae. aegypti (22). In the present article we demonstrate that the 15 15 15 mosquitoes incorporate N from NH4Cl into [5- N]-glutamine nitrogen of the amide group of Gln contributes to uric acid and use the 15N of the amide group of glutamine to produce synthesis in mosquitoes and, surprisingly, that uric acid can be 15 labeled uric acid. -

Francisella Tularensis 6/06 Tularemia Is a Commonly Acquired Laboratory Colony Morphology Infection; All Work on Suspect F
Francisella tularensis 6/06 Tularemia is a commonly acquired laboratory Colony Morphology infection; all work on suspect F. tularensis cultures .Aerobic, fastidious, requires cysteine for growth should be performed at minimum under BSL2 .Grows poorly on Blood Agar (BA) conditions with BSL3 practices. .Chocolate Agar (CA): tiny, grey-white, opaque A colonies, 1-2 mm ≥48hr B .Cysteine Heart Agar (CHA): greenish-blue colonies, 2-4 mm ≥48h .Colonies are butyrous and smooth Gram Stain .Tiny, 0.2–0.7 μm pleomorphic, poorly stained gram-negative coccobacilli .Mostly single cells Growth on BA (A) 48 h, (B) 72 h Biochemical/Test Reactions .Oxidase: Negative A B .Catalase: Weak positive .Urease: Negative Additional Information .Can be misidentified as: Haemophilus influenzae, Actinobacillus spp. by automated ID systems .Infective Dose: 10 colony forming units Biosafety Level 3 agent (once Francisella tularensis is . Growth on CA (A) 48 h, (B) 72 h suspected, work should only be done in a certified Class II Biosafety Cabinet) .Transmission: Inhalation, insect bite, contact with tissues or bodily fluids of infected animals .Contagious: No Acceptable Specimen Types .Tissue biopsy .Whole blood: 5-10 ml blood in EDTA, and/or Inoculated blood culture bottle Swab of lesion in transport media . Gram stain Sentinel Laboratory Rule-Out of Francisella tularensis Oxidase Little to no growth on BA >48 h Small, grey-white opaque colonies on CA after ≥48 h at 35/37ºC Positive Weak Negative Positive Catalase Tiny, pleomorphic, faintly stained, gram-negative coccobacilli (red, round, and random) Perform all additional work in a certified Class II Positive Biosafety Cabinet Weak Negative Positive *Oxidase: Negative Urease *Catalase: Weak positive *Urease: Negative *Oxidase, Catalase, and Urease: Appearances of test results are not agent-specific. -

Nitrilase 1 Modulates Lung Tumor Progression in Vitro and in Vivo
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 7, No. 16 Nitrilase 1 modulates lung tumor progression in vitro and in vivo Yong Antican Wang1, Yunguang Sun2,5, Justin M. Le Blanc1, Charalambos Solomides3, Tingting Zhan4, Bo Lu1 1Department of Radiation Oncology, Thomas Jefferson University, Philadelphia, PA, 19107, USA 2Department of Cancer Biology, Thomas Jefferson University, Philadelphia, PA, 19107, USA 3Department of Pathology, Thomas Jefferson University, Philadelphia, PA, 19107, USA 4Department of Pharmacology, Thomas Jefferson University, Philadelphia, PA, 19107, USA 5Department of Pathology, Medical College of Wisconsin, Milwaukee, WI, 53226, USA Correspondence to: Bo Lu, e-mail: [email protected]. Keywords: NIT1, lung cancer, KRAS, NSCLC, tumor suppressor Received: October 01, 2015 Accepted: January 23, 2016 Published: March 10, 2016 ABSTRACT Uncovering novel growth modulators for non-small cell lung cancer (NSCLC) may lead to new therapies for these patients. Previous studies suggest Nit1 suppresses chemically induced carcinogenesis of the foregut in a mouse model. In this study we aimed to determine the role of Nit1 in a transgenic mouse lung cancer model driven by a G12D Kras mutation. Nit1 knockout mice (Nit1−/−) were crossed with KrasG12D/+ mice to investigate whether a G12D Kras mutation and Nit1 inactivation interact to promote or inhibit the development of NSCLC. We found that lung tumorigenesis was suppressed in the Nit1-null background (Nit1−/−:KrasG12D/+). Micro-CT scans and gross tumor measurements demonstrated a 5-fold reduction in total tumor volumes compared to Nit1+/+KrasG12D/+ (p<0.01). Furthermore, we found that Nit1 is highly expressed in human lung cancer tissues and cell lines and use of siRNA against Nit1 decreased overall cell survival of lung cancer cells in culture. -

Purification and Characterization of a Novel Nitrilase of Rhodococcus
JOURNAL OF BACTERIOLOGY, Sept. 1990, p. 4807-4815 Vol. 172, No. 9 0021-9193/90/094807-09$02.00/0 Copyright X3 1990, American Society for Microbiology Purification and Characterization of a Novel Nitrilase of Rhodococcus rhodochrous K22 That Acts on Aliphatic Nitriles MICHIHIKO KOBAYASHI,* NORIYUKI YANAKA, TORU NAGASAWA, AND HIDEAKI YAMADA Department ofAgricultural Chemistry, Faculty ofAgriculture, Kyoto University, Sakyo-ku, Kyoto 606, Japan Received 18 April 1990/Accepted 8 June 1990 A novel nitrilase that preferentially catalyzes the hydrolysis of aliphatic nitriles to the corresponding carboxylic acids and ammonia was found in the cells of a facultative crotononitrile-utilizing actinomycete isolated from soil. The strain was taxonomically studied and identified as Rhodococcus rhodochrous. The nitrilase was purified, with 9.08% overall recovery, through five steps from a cell extract of the stain. After the last step, the purified enzyme appeared to be homogeneous, as judged by polyacrylamide gel electrophoresis, analytical centrifugation, and double immunodiffusion in agarose. The relative molecular weight values for the native enzyme, estimated from the ultracentrifugal equilibrium and by high-performance liquid chromatog- raphy, were approximately 604,000 + 30,000 and 650,000, respectively, and the enzyme consisted of 15 to 16 subunits identical in molecular weight (41,000). The enzyme acted on aliphatic olefinic nitriles such as crotononitrile and acrylonitrile as the most suitable substrates. The apparent Km values for crotononitrile and acrylonitrile were 18.9 and 1.14 mM, respectively. The nitrilase also catalyzed the direct hydrolysis of saturated aliphatic nitriles, such as valeronitrile, 4-chlorobutyronitrile, and glutaronitrile, to the correspond- ing acids without the formation of amide intermediates. -

Extended Spectrum Β-Lactamases and Class C Β
F1000Research 2020, 9:774 Last updated: 01 APR 2021 RESEARCH ARTICLE Extended spectrum β-lactamases and class C β-lactamases gene frequency in Pseudomonas aeruginosa isolated from various clinical specimens in Khartoum State, Sudan: a cross sectional study [version 1; peer review: 1 not approved] Dina N. Abdelrahman 1,2, Aya A. Taha2, Mazar M. Dafaallah2, Alaa Abdelgafoor Mohammed3, Abdel Rahim M. El Hussein 1, Ahmed I. Hashim 2, Yousif F. Hamedelnil2, Hisham N. Altayb 4 1Department of Virology, Central Laboratory, Khartoum, Sudan 2Department of Microbiology, College of Medical Laboratory Sciences, Sudan University of Science and Technology, Khartoum, Sudan 3Department of Pharmaceutical Biotechnology, College of Pharmacy, Ahfad University for Women, Omdurman, Khartoum, Sudan 4Biochemistry Department, Faculty of Sciences, King Abdulaziz University, Jeddah, Saudi Arabia v1 First published: 27 Jul 2020, 9:774 Open Peer Review https://doi.org/10.12688/f1000research.24818.1 Second version: 15 Sep 2020, 9:774 https://doi.org/10.12688/f1000research.24818.2 Reviewer Status Latest published: 01 Apr 2021, 9:774 https://doi.org/10.12688/f1000research.24818.3 Invited Reviewers 1 2 Abstract Background: Pseudomonas aeruginosa is a pathogenic bacterium, version 3 causing nosocomial infections with intrinsic and acquired resistance (revision) mechanisms to a large group of antibiotics, including β-lactams. This 01 Apr 2021 study aimed to determine the susceptibility pattern to selected antibiotics and to index the first reported β-lactamases gene version 2 (extended spectrum β-lactamases (ESBLs) genes and class C β- (revision) report report lactamases genes) frequency in Ps. aeruginosa in Khartoum State, 15 Sep 2020 Sudan. Methods: 121 Ps. -

Direct Link to Fulltext
ISOLATION, IDENTIFICATION AND SUBSTRATE SPECIFICITY OF A NITRILASE PRODUCING BACTERIA, Acidovorax sp. SK1 Soumya Koippully, Subha Swaraj Pattnaik, Siddhardha Busi* Address(es): Department of Microbiology, School of Life Sciences, Pondicherry University, Puducherry-605 014, India. *Corresponding author: [email protected] doi: 10.15414/jmbfs.2018.8.2.788-793 ARTICLE INFO ABSTRACT Received 13. 5. 2018 The process of biocatalysis or biotransformation remains the core of industrial biotechnology owing to its importance in the synthesis of Revised 3. 9. 2018 high-value products in a cost effective manner. Nitrile biotransformation has also achieved considerable attention in last few decades Accepted 5. 9. 2018 due to its widespread biotechnological and industrial applications. In the present study, a versatile, potent nitrile-degrading bacterium, Published 1. 10. 2018 Acidovorax sp. SK1 was isolated from the cracker waste dumping site of Sivakasi, Tamilnadu and characterized for its biocatalytic potential, nitrilase production and enzyme activity. A pH sensitive indicator-based assay was performed to identify the nitrile degrading ability of the isolated samples. Semi quantitative High performance thin layer chromatographic (HPTLC) method was performed for Regular article quantitative measurement of mandelic acid produced from degradation of nitrile compound, mandelonitrile. The optimization of medium and nutritional parameters were studied for the improvement of nitrilase activity, which indicated that maximum nitrilase activity was observed at an optimum pH of 7.0, agitation at 100 rpm, glucose as best carbon source (10 g/L) and yeast extract (0.1 g/L) as principal nitrogen source. Biomass is also a critical parameter in the biocatalysis of mandelonitrile to mandelic acid and at a biomass of 100 mg/L, maximum nitrilase activity of 0.026 I.U was observed. -

(12) United States Patent (10) Patent No.: US 9.422,609 B2 Teichberg (45) Date of Patent: Aug
USOO9422609B2 (12) United States Patent (10) Patent No.: US 9.422,609 B2 Teichberg (45) Date of Patent: Aug. 23, 2016 (54) METHODS, COMPOSITIONS AND DEVICES (58) Field of Classification Search FOR MANTAINING CHEMICAL BALANCE CPC ........................ C02F 1/725; C12Y 305/01005 OF CHLORINATED WATER USPC ........................... 210/754; 435/195, 227 231 See application file for complete search history. (75) Inventor: Vivian I. Teichberg, Savyon (IL) (56) References Cited (73) Assignees: Mia Levite, Savyon (IL); Yaar Teichberg, Savyon (IL); Nof Lyle U.S. PATENT DOCUMENTS Teichberg, Savyon (IL) 4,793,935 A * 12/1988 Stillman ............... CO2F 1.5236 21Of727 (*) Notice: Subject to any disclaimer, the term of this 6,673,582 B2 * 1/2004 McTavish ..................... 435/122 patent is extended or adjusted under 35 U.S.C. 154(b) by 1044 days. (Continued) (21) Appl. No.: 12/225.18O FOREIGN PATENT DOCUMENTS y x- - - 9 AU 41971 5, 1979 (22) PCT Filed: Mar. 14, 2007 GB 2025919 1, 1980 (86). PCT No.: PCT/L2007/OOO336 (Continued) S 371 (c)(1) OTHER PUBLICATIONS (2), (4) Date: Sep. 16, 2008 Examiner's Report Dated Oct. 6, 2010 From the Australian Govern (87) PCT Pub. No.: WO2007/107.981 ment, IP Australia Re. Application No. 2007228391. (Continued) PCT Pub. Date: Sep. 27, 2007 (65) Prior Publication Data Primary Examiner — Peter Keyworth (74) Attorney, Agent, or Firm — Browdy and Neimark, US 201OfO270228A1 Oct. 28, 2010 PLLC Related U.S. Application Data (57) ABSTRACT (60) Provisional application No. 60/783,028, filed on Mar. A composition-of-matter for use in water treatment, com 17, 2006.