Interim Report IR-11-026 Repeated Unidirectional Introgression Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ophthalmotilapia Boops (BOULENGER, 1901)
DCG_Info_05_2016_HR_20160420_Karlsson_ohne_Haustein_DCG_Info 20.04.2016 06:36 Seite 106 Ophthalmotilapia boops (BOULENGER, 1901) Wolfgang Staeck Abb. 1. Männchen von Ophthalmotilapia boops über seiner Laichgrube auf einem Felsen bei Kampemba Point (Kleines Bild: Ophthalmotilapia boops und Tropheus bei Kampemba Point) Meine erste Begegnung mit diesem un- Beide sprachen kein Englisch, sondern geografische Farbvariante von Oph- gewöhnlichen Tanganjikasee-Bunt- unterhielten sich gewöhnlich auf Sua- thalmotilapia ventralis hielt, zu fangen, barsch liegt nun bereits fast vierzig heli, das ich nicht beherrschte. Da in weil wir nicht über die dazu benötigte Jahre zurück. Im Jahre 1977 bereiste Burundi aber neben der afrikanischen Ausrüstung verfügten, um sie heil bis ich zusammen mit zwei Fängern des Muttersprache auch Französisch ge- in das weit entfernte Kigoma zu trans- damals in Dar es Salaam lebenden Ex- sprochen wurde, war die Kommunika- portieren. porteurs Misha Fainzilber in einem der tion in unserer kleinen Reisegruppe dort üblichen motorisierten Fischer- kein unlösbares Problem. Systematik und Merkmale von Oph- boote von Kigoma aus die zu Tansania thalmotilapia gehörende Ostküste des Tanganjikasees Ich erinnere mich noch genau über in südlicher Richtung bis nach Kipili. meine Begeisterung, als ich in der Um- Ophthalmotilapia boops gehört zu den Ziel unserer fast drei Wochen dauern- gebung der Ortschaft Kipili schnor- ersten Arten, die aus dem Tanganjika- den Entdeckungsreise war es, für den chelte und dort im Uferbereich der see beschrieben wurden, denn die bei- Exporteur eine erste Bestandsaufnahme Insel Kerenge erstmals die auffallend den Typusexemplare sind von dem der in diesem Gebiet vorkommenden kontrastreich tief schwarz und leuch- englischen Forschungsreisenden MOORE Buntbarsche vorzunehmen. tend hellblau gezeichneten Männchen bereits gegen Ende des 19. -

And Giant Guitarfish (Rhynchobatus Djiddensis)
VIRAL DISCOVERY IN BLUEGILL SUNFISH (LEPOMIS MACROCHIRUS) AND GIANT GUITARFISH (RHYNCHOBATUS DJIDDENSIS) BY HISTOPATHOLOGY EVALUATION, METAGENOMIC ANALYSIS AND NEXT GENERATION SEQUENCING by JENNIFER ANNE DILL (Under the Direction of Alvin Camus) ABSTRACT The rapid growth of aquaculture production and international trade in live fish has led to the emergence of many new diseases. The introduction of novel disease agents can result in significant economic losses, as well as threats to vulnerable wild fish populations. Losses are often exacerbated by a lack of agent identification, delay in the development of diagnostic tools and poor knowledge of host range and susceptibility. Examples in bluegill sunfish (Lepomis macrochirus) and the giant guitarfish (Rhynchobatus djiddensis) will be discussed here. Bluegill are popular freshwater game fish, native to eastern North America, living in shallow lakes, ponds, and slow moving waterways. Bluegill experiencing epizootics of proliferative lip and skin lesions, characterized by epidermal hyperplasia, papillomas, and rarely squamous cell carcinoma, were investigated in two isolated poopulations. Next generation genomic sequencing revealed partial DNA sequences of an endogenous retrovirus and the entire circular genome of a novel hepadnavirus. Giant Guitarfish, a rajiform elasmobranch listed as ‘vulnerable’ on the IUCN Red List, are found in the tropical Western Indian Ocean. Proliferative skin lesions were observed on the ventrum and caudal fin of a juvenile male quarantined at a public aquarium following international shipment. Histologically, lesions consisted of papillomatous epidermal hyperplasia with myriad large, amphophilic, intranuclear inclusions. Deep sequencing and metagenomic analysis produced the complete genomes of two novel DNA viruses, a typical polyomavirus and a second unclassified virus with a 20 kb genome tentatively named Colossomavirus. -

Spatial Models of Speciation 1.0Cm Modelos Espaciais De Especiação
UNIVERSIDADE ESTADUAL DE CAMPINAS INSTITUTO DE BIOLOGIA CAROLINA LEMES NASCIMENTO COSTA SPATIAL MODELS OF SPECIATION MODELOS ESPACIAIS DE ESPECIAÇÃO CAMPINAS 2019 CAROLINA LEMES NASCIMENTO COSTA SPATIAL MODELS OF SPECIATION MODELOS ESPACIAIS DE ESPECIAÇÃO Thesis presented to the Institute of Biology of the University of Campinas in partial fulfill- ment of the requirements for the degree of Doc- tor in Ecology Tese apresentada ao Instituto de Biologia da Universidade Estadual de Campinas como parte dos requisitos exigidos para a obtenção do título de Doutora em Ecologia Orientador: Marcus Aloizio Martinez de Aguiar ESTE ARQUIVO DIGITAL CORRESPONDE À VERSÃO FINAL DA TESE DEFENDIDA PELA ALUNA CAROLINA LEMES NASCIMENTO COSTA, E ORIENTADA PELO PROF DR. MAR- CUS ALOIZIO MARTINEZ DE AGUIAR. CAMPINAS 2019 Ficha catalográfica Universidade Estadual de Campinas Biblioteca do Instituto de Biologia Mara Janaina de Oliveira - CRB 8/6972 Costa, Carolina Lemes Nascimento, 1989- C823s CosSpatial models of speciation / Carolina Lemes Nascimento Costa. – Campinas, SP : [s.n.], 2019. CosOrientador: Marcus Aloizio Martinez de Aguiar. CosTese (doutorado) – Universidade Estadual de Campinas, Instituto de Biologia. Cos1. Especiação. 2. Radiação adaptativa (Evolução). 3. Modelos biológicos. 4. Padrão espacial. 5. Macroevolução. I. Aguiar, Marcus Aloizio Martinez de, 1960-. II. Universidade Estadual de Campinas. Instituto de Biologia. III. Título. Informações para Biblioteca Digital Título em outro idioma: Modelos espaciais de especiação Palavras-chave em inglês: Speciation Adaptive radiation (Evolution) Biological models Spatial pattern Macroevolution Área de concentração: Ecologia Titulação: Doutora em Ecologia Banca examinadora: Marcus Aloizio Martinez de Aguiar [Orientador] Mathias Mistretta Pires Sabrina Borges Lino Araujo Rodrigo André Caetano Gustavo Burin Ferreira Data de defesa: 25-02-2019 Programa de Pós-Graduação: Ecologia Powered by TCPDF (www.tcpdf.org) Comissão Examinadora: Prof. -

Exchange April 2018 Area of Concern—Lake Tanganyika Do You CARE
The CARES April 2018 Exchange Area of Concern—Lake Tanganyika Do You CARE Crossword Challenge Data Submission Deadline April 30 Welcome to The CARES Exchange. The primary intent of this publication is to make available a listing of CARES fish from the CARES membership to those that may be searching for CARES species. The Cichlid Room Companion is the most It is important to understand that all transac- comprehensive website for reliable cichlid tions are between the buyer and seller and information in the world. For all things cich- CARES in no way moderates any exchanges lid, including information, photos, and videos including shipping problems, refunds, or bad on most CARES Priority List species, visit blood between the two parties. This directo- CRC at www.cichlidae.com. ry merely provides an avenue to which CARES fish may be located. As with all sales, be certain that all the elements of the The CARES Family exchange are worked out before purchasing American Cichlid Association or shipping. Aquarium Club of Lancaster County Brooklyn Aquarium Society No hybrids will knowingly be listed. Capital Cichlid Association Chatham-Kent Aquarium Society There is no cost to place a for sale ad. Your Cichlid Club of York ad may be submitted by contacting the editor, Columbus Area Fish Enthusiasts Greg Steeves, at [email protected]. Danbury Area Aquarium Society Durham Region Aquarium Society If your organization is interested in partici- Federation of Texas Aquarium Societies pating in CARES, review the CARES Startup Grand Valley Aquarium Club tab on the website CARESforfish.org, then Greater Cincinnati Aquarium Society contact Klaus Steinhaus at Greater City Aquarium Society [email protected]. -

The Initial Response of Females Towards Congeneric Males Matches the Propensity to Hybridize
bioRxiv preprint doi: https://doi.org/10.1101/2021.08.07.455508; this version posted August 8, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 The initial response of females towards congeneric males matches the propensity to hybridize 2 in Ophthalmotilapia. 3 4 Maarten Van Steenberge1,2, Noémie Jublier3,4, Loïc Kéver3, Sophie Gresham1,5, Sofie 5 Derycke1,6, Jos Snoeks2,7, Eric Parmentier3, Pascal Poncin4, Erik Verheyen1,5 6 7 1. Operational Directorate Taxonomy and phylogeny, Royal Belgian Institute of Natural 8 Sciences, 1000 Brussels, Belgium 9 2. Laboratory of Biodiversity and Evolutionary Genomics, KU Leuven, Charles 10 Deberiotstraat 32, 3000 Leuven, Belgium 11 3. Laboratory of Functional and Evolutionary Biology, FOCUS, Liège University, 4000 12 Liège, Belgium 13 4. Laboratory of Fish and Amphibian Ethology, Behavioural Biology Unit, FOCUS, 14 Liège University, 4000 Liège, Belgium 15 5. Department Biology, Evolutionary Ecology, University of Antwerp, 2610 Antwerp, 16 Belgium 17 6. Flanders research institute for agriculture, fisheries and food, Animal Sciences Unit – 18 Aquatic Environment and Quality, Ankerstraat 1, 8400 Oostende, Belgium. 19 7. Section Vertebrates, Ichthyology, Royal Museum for Central Africa, 3080 Tervuren, 20 Belgium 21 22 Corresponding author: Maarten Van Steenberge ([email protected]) 23 24 Key words: 25 Species recognition, mate choice, behaviour, Lake Tanganyika, Africa 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.08.07.455508; this version posted August 8, 2021. -

Eine Neue Geografische Farbvariante Von Ophthalmotilapia Sp. „Whitecap“
Die neue Farbvariante von Ophthalmotilapia sp. „Whitecap“ im flachen Wasser bei der Ortschaft Ninde. Eine neue geografische Farbvariante von Ophthalmotilapia sp. „Whitecap“ Text: Wolfgang Staeck Fotos: Magnus und Mikael Karlsson Zu den bemerkenswertesten Buntbar- dieser Buntbarsche in ungewöhnlicher gelegen, wurde diese Farbform, die die schen des Tanganjikasees zählen wegen Weise ver längert und fadenartig ausge- südlichste Population von Ophthalmo- ihrer prächtigen Färbung und unge- zogen sind. Wahrscheinlich existieren tilapia sp. „Whitecap“ ist, nicht mehr wöhnlichen Flossenform zweifellos die aber auch noch unbeschriebene Arten. gefunden. Vertreter der Gattung Ophthalmotila- pia, die von PELLEGRIN bereits im Jahre Die jetzt erstmals im Foto vorgestellte Nach den Beobachtungen der Entde- 1904 beschrieben wurde. POLL über- Farbform von Ophthalmotilapia sp. cker haben die Fische eine ungewöhn- führte später einen Teil der Arten „Whitecap“ wurde von Magnus und lich geringe Individuendichte und sind wegen ihrer ausschließlich kegelförmi- Mikael KARLSSON schon im Jahr 2008 deshalb an den meisten Fundorten sel- gen Zähne, die nicht dreispitzig sind, entdeckt, als sie an der zu Tansania ge- ten. KARLSSON & KARLSSON führen sondern nur eine Spitze haben, in die hörenden südlichen Küste des Tanga- von ihm neu aufgestellte Gattung Oph- njikasees eine Dokumentation der thalmochromis (1956), was jedoch Verbreitungsgrenzen der dort vorkom- LIEM (1981) in einer Revision dieses menden Buntbarsche durchführten Verwandtschaftskreises wieder rück- (KARLSSON & KARLSSON 2013). Die gängig machte und die Gattung zum neue Farbvariante kommt südlich von jüngeren Synonym für Ophthalmotila- Kipili an einem nur 16 Kilometer lan- pia erklärte. Nach POLL (1986) enthält gen Küstenstreifen zwischen Pinga Ophthalmotilapia vier Arten, die in der Point (nahe der Ortschaft Mkilinga) Aquaristik unter der Bezeichnung Fa- und Chisanze Point (nördlich der Nordseite des Dorfes Ninde Village. -

The Use of Different Sensory Modalities in Predator Discrimination
Behav Ecol Sociobiol (2017) 71: 143 DOI 10.1007/s00265-017-2371-8 ORIGINAL ARTICLE Smell or vision? The use of different sensory modalities in predator discrimination Stefan Fischer1,2,3 & Evelyne Oberhummer1 & Filipa Cunha-Saraiva1,4 & Nina Gerber1,5 & Barbara Taborsky1 Received: 6 November 2016 /Revised: 19 August 2017 /Accepted: 28 August 2017 /Published online: 8 September 2017 # The Author(s) 2017. This article is an open access publication Abstract to a standardized threat stimulus combined with either preda- Theory predicts that animals should adjust their escape re- tor or non-predator cues presented either visually or chemical- sponses to the perceived predation risk. The information ani- ly. We predicted that flight responses towards a threat stimulus mals obtain about potential predation risk may differ qualita- are more pronounced if cues of dangerous rather than harm- tively depending on the sensory modality by which a cue is less heterospecifics are presented and that N. pulcher,beingan perceived. For instance, olfactory cues may reveal better in- aquatic species, relies more on olfaction when discriminating formation about the presence or absence of threats, whereas between dangerous and harmless heterospecifics. N. pulcher visual information can reliably transmit the position and po- responded faster to the threat stimulus, reached a refuge faster tential attack distance of a predator. While this suggests a and entered a refuge more likely when predator cues were differential use of information perceived through the two sen- perceived. Unexpectedly, the sensory modality used to per- sory channels, the relative importance of visual vs. olfactory ceive the cues did not affect the escape response or the dura- cues when distinguishing between different predation threats tion of the recovery phase. -

New Fossils of Cichlids from the Miocene of Kenya and Clupeids from the Miocene of Greece (Teleostei)
The importance of articulated skeletons in the identification of extinct taxa: new fossils of cichlids from the Miocene of Kenya and clupeids from the Miocene of Greece (Teleostei) Dissertation zur Erlangung des Doktorgrades an der Fakultät für Geowissenschaften der Ludwig-Maximilians-Universität München Vorgelegt von Charalampos Kevrekidis München, 28. September 2020 Erstgutacher: Prof. Dr. Bettina Reichenbacher Zweitgutacher: PD Dr. Gertrud Rößner Tag der mündlichen Prüfung: 08.02.2021 2 Statutory declaration and statement I hereby confirm that my Thesis entitled “Fossil fishes from terrestrial sediments of the Miocene of Africa and Europe”, is the result of my own original work. Furthermore, I certify that this work contains no material which has been accepted for the award of any other degree or diploma in my name, in any university and, to the best of my knowledge and belief, contains no material previously published or written by another person, except where due reference has been made in the text. In addition, I certify that no part of this work will, in the future, be used in a submission in my name, for any other degree or diploma in any university or other tertiary institution without the prior approval of the Ludwig-Maximilians-Universität München. München, 21.09.2020 Charalampos Kevrekidis 3 Abstract Fishes are important components of aquatic faunas, but our knowledge on the fossil record of some taxa, relative to their present diversity, remains poor. This can be due to a rarity of such fossils, as is the case for the family Cichlidae (cichlids). Another impediment is the rarity of well-preserved skeletons of fossil fishes. -

Testing the Potential of Environmental DNA Methods for Surveying Lake Tanganyika's Highly Diverse Fish Communities Christopher J
Testing the potential of environmental DNA methods for surveying Lake Tanganyika's highly diverse fish communities Christopher James Doble A thesis submitted for the degree of Doctor of Philosophy Department of Genetics, Evolution and Environment University College London April 2020 1 Declaration I, Christopher James Doble, confirm the work presented in this thesis is my own. Where information has been derived from other sources, I confirm this has been indicated in the thesis. Christopher James Doble Date: 27/04/2020 2 Statement of authorship I planned and undertook fieldwork to the Kigoma region of Lake Tanganyika, Tanzania in 2016 and 2017. This included obtaining research permits, collecting environmental DNA samples and undertaking fish community visual survey data used in Chapters three and four. For Chapter two, cichlid reference database sequences were sequenced by Walter Salzburger’s research group at the University of Basel. I extracted required regions from mitochondrial genome alignments during a visit to Walter’s research group. Other reference sequences were obtained by Sanger sequencing. I undertook the DNA extractions and PCR amplifications for all samples, with the clean-up and sequencing undertaken by the UCL Sequencing facility. I undertook the method development, DNA extractions, PCR amplifications and library preparations for each of the next generation sequencing runs in Chapters three and four at the NERC Biomolecular Analysis Facility Sheffield. Following training by Helen Hipperson at the NERC Biomolecular Analysis Facility in Sheffield, I undertook the bioinformatic analysis of sequence data in Chapters three and four. I also carried out all the data analysis within each chapter. Chapters two, three and parts of four have formed a manuscript recently published in Environmental DNA (Doble et al. -
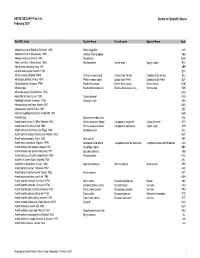
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

Diversity, Origin and Intra- Specific Variability
Contributions to Zoology, 87 (2) 105-132 (2018) Monogenean parasites of sardines in Lake Tanganyika: diversity, origin and intra- specific variability Nikol Kmentová1, 15, Maarten Van Steenberge2,3,4,5, Joost A.M. Raeymaekers5,6,7, Stephan Koblmüller4, Pascal I. Hablützel5,8, Fidel Muterezi Bukinga9, Théophile Mulimbwa N’sibula9, Pascal Masilya Mulungula9, Benoît Nzigidahera†10, Gaspard Ntakimazi11, Milan Gelnar1, Maarten P.M. Vanhove1,5,12,13,14 1 Department of Botany and Zoology, Faculty of Science, Masaryk University, Kotlářská 2, 611 37 Brno, Czech Republic 2 Biology Department, Royal Museum for Central Africa, Leuvensesteenweg 13, 3080, Tervuren, Belgium 3 Operational Directorate Taxonomy and Phylogeny, Royal Belgian Institute of Natural Sciences, Vautierstraat 29, B-1000 Brussels, Belgium 4 Institute of Biology, University of Graz, Universitätsplatz 2, A-8010 Graz, Austria 5 Laboratory of Biodiversity and Evolutionary Genomics, Department of Biology, University of Leuven, Ch. Deberiotstraat 32, B-3000 Leuven, Belgium 6 Centre for Biodiversity Dynamics, Department of Biology, Norwegian University of Science and Technology, N-7491 Trondheim, Norway 7 Faculty of Biosciences and Aquaculture, Nord University, N-8049 Bodø, Norway 8 Flanders Marine Institute, Wandelaarkaai 7, 8400 Oostende, Belgium 9 Centre de Recherche en Hydrobiologie, Département de Biologie, B.P. 73 Uvira, Democratic Republic of Congo 10 Office Burundais pour la Protection de l‘Environnement, Centre de Recherche en Biodiversité, Avenue de l‘Imprimerie Jabe 12, B.P. -

Functional Coupling Constrains Craniofacial Diversification in Lake
Downloaded from http://rsbl.royalsocietypublishing.org/ on July 3, 2017 Evolutionary biology Functional coupling constrains craniofacial rsbl.royalsocietypublishing.org diversification in Lake Tanganyika cichlids Masahito Tsuboi1, Alejandro Gonzalez-Voyer2,3 and Niclas Kolm3 1Evolutionary Biology Centre, Department of Ecology and Genetics/Animal Ecology, Uppsala University, Research Norbyva¨gen 18D, 75236 Uppsala, Sweden 2Laboratorio de Conducta Animal, Instituto de Ecologı´a, Universidad Nacional Auto´noma de Me´xico, Cite this article: Tsuboi M, Gonzalez-Voyer A, Circuito Exterior S/N, Ciudad Universitaria, UNAM, D.F. 04510, Me´xico 3 Kolm N. 2015 Functional coupling constrains Department of Zoology/Ethology, Stockholm University, Svante Arrhenius va¨g 18B, 10691 Stockholm, Sweden craniofacial diversification in Lake Tanganyika MT, 0000-0002-0144-2893 cichlids. Biol. Lett. 11: 20141053. http://dx.doi.org/10.1098/rsbl.2014.1053 Functional coupling, where a single morphological trait performs multiple func- tions, is a universal feature of organismal design. Theory suggests that functional coupling may constrain the rate of phenotypic evolution, yet empirical tests of this hypothesis are rare. In fish, the evolutionary transition from guarding the Received: 11 December 2014 eggs on a sandy/rocky substrate (i.e. substrate guarding) to mouthbrood- ing introduces a novel function to the craniofacial system and offers an ideal Accepted: 9 April 2015 opportunity to test the functional coupling hypothesis. Using a combination of geometric morphometrics and a recently developed phylogenetic compara- tive method, we found that head morphology evolution was 43% faster in substrate guarding species than in mouthbrooding species. Furthermore, for Subject Areas: species in which females were solely responsible for mouthbrooding the evolution, ecology, behaviour males had a higher rate of head morphology evolution than in those with bi- parental mouthbrooding.