Complete Chloroplast Genome Sequence of MD-2 Pineapple and Its Comparative Analysis Among Nine Other Plants from the Subclass Commelinidae R
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Duplication and Diversification of the LEAFY HULL STERILE1 and Oryza
Christensen and Malcomber EvoDevo 2012, 3:4 http://www.evodevojournal.com/content/3/1/4 RESEARCH Open Access Duplication and diversification of the LEAFY HULL STERILE1 and Oryza sativa MADS5 SEPALLATA lineages in graminoid Poales Ashley R Christensen1,2 and Simon T Malcomber1* Abstract Background: Gene duplication and the subsequent divergence in function of the resulting paralogs via subfunctionalization and/or neofunctionalization is hypothesized to have played a major role in the evolution of plant form. The LEAFY HULL STERILE1 (LHS1) SEPALLATA (SEP) genes have been linked with the origin and diversification of the grass spikelet, but it is uncertain 1) when the duplication event that produced the LHS1 clade and its paralogous lineage Oryza sativa MADS5 (OSM5) occurred, and 2) how changes in gene structure and/or expression might have contributed to subfunctionalization and/or neofunctionalization in the two lineages. Methods: Phylogenetic relationships among 84 SEP genes were estimated using Bayesian methods. RNA expression patterns were inferred using in situ hybridization. The patterns of protein sequence and RNA expression evolution were reconstructed using maximum parsimony (MP) and maximum likelihood (ML) methods, respectively. Results: Phylogenetic analyses mapped the LHS1/OSM5 duplication event to the base of the grass family. MP character reconstructions estimated a change from cytosine to thymine in the first codon position of the first amino acid after the Zea mays MADS3 (ZMM3) domain converted a glutamine to a stop codon in the OSM5 ancestor following the LHS1/OSM5 duplication event. RNA expression analyses of OSM5 co-orthologs in Avena sativa, Chasmanthium latifolium, Hordeum vulgare, Pennisetum glaucum, and Sorghum bicolor followed by ML reconstructions of these data and previously published analyses estimated a complex pattern of gain and loss of LHS1 and OSM5 expression in different floral organs and different flowers within the spikelet or inflorescence. -

Grass Genera in Townsville
Grass Genera in Townsville Nanette B. Hooker Photographs by Chris Gardiner SCHOOL OF MARINE and TROPICAL BIOLOGY JAMES COOK UNIVERSITY TOWNSVILLE QUEENSLAND James Cook University 2012 GRASSES OF THE TOWNSVILLE AREA Welcome to the grasses of the Townsville area. The genera covered in this treatment are those found in the lowland areas around Townsville as far north as Bluewater, south to Alligator Creek and west to the base of Hervey’s Range. Most of these genera will also be found in neighbouring areas although some genera not included may occur in specific habitats. The aim of this book is to provide a description of the grass genera as well as a list of species. The grasses belong to a very widespread and large family called the Poaceae. The original family name Gramineae is used in some publications, in Australia the preferred family name is Poaceae. It is one of the largest flowering plant families of the world, comprising more than 700 genera, and more than 10,000 species. In Australia there are over 1300 species including non-native grasses. In the Townsville area there are more than 220 grass species. The grasses have highly modified flowers arranged in a variety of ways. Because they are highly modified and specialized, there are also many new terms used to describe the various features. Hence there is a lot of terminology that chiefly applies to grasses, but some terms are used also in the sedge family. The basic unit of the grass inflorescence (The flowering part) is the spikelet. The spikelet consists of 1-2 basal glumes (bracts at the base) that subtend 1-many florets or flowers. -

Understory Community Shifts in Response to Repeated Fire and Fire Surrogate Treatments in the Southern Appalachian Mountains, USA Emily C
Oakman et al. Fire Ecology (2021) 17:7 Fire Ecology https://doi.org/10.1186/s42408-021-00097-1 ORIGINAL RESEARCH Open Access Understory community shifts in response to repeated fire and fire surrogate treatments in the southern Appalachian Mountains, USA Emily C. Oakman1, Donald L. Hagan1* , Thomas A. Waldrop2 and Kyle Barrett1 Abstract Background: Decades of fire exclusion in the southern Appalachian Mountains, USA, has led to changing forest structure and species composition over time. Forest managers and scientists recognize this and are implementing silvicultural treatments to restore forest communities. In this study, conducted at the southern Appalachian Fire and Fire Surrogate Study site in Green River Game Land, North Carolina, USA, we assessed the effects of four fuel- reduction methods (burned four times, B; mechanical treatment two times, M; mechanical treatment two times plus burned four times, MB; and control, C) on the changes in understory community from pre-treatment to post- treatment years (2001 to 2016). We used non-metric multidimensional scaling (NMDS) to determine overall understory community heterogeneity, agglomerative hierarchical cluster analyses (AHCA) to determine finer-scale changes in understory community structure, and indicator species analyses (ISA) to identify the species that were associated with the different fuel reduction treatments over time. Results: The NMDS ordination showed little separation between treatment polygons. The AHCA resulted in two main categories of understory species responses based on how treatment plots clustered together: (1) species apparently unaffected by the treatments (i.e., no treatment pattern present within cluster); and (2) species that responded to B, M, or MB treatments (i.e., pattern of treatment plots present within cluster). -

Flora and Vegetation Survey of the Proposed Kwinana to Australind Gas
__________________________________________________________________________________ FLORA AND VEGETATION SURVEY OF THE PROPOSED KWINANA TO AUSTRALIND GAS PIPELINE INFRASTRUCTURE CORRIDOR Prepared for: Bowman Bishaw Gorham and Department of Mineral and Petroleum Resources Prepared by: Mattiske Consulting Pty Ltd November 2003 MATTISKE CONSULTING PTY LTD DRD0301/039/03 __________________________________________________________________________________ TABLE OF CONTENTS Page 1. SUMMARY............................................................................................................................................... 1 2. INTRODUCTION ..................................................................................................................................... 2 2.1 Location................................................................................................................................................. 2 2.2 Climate .................................................................................................................................................. 2 2.3 Vegetation.............................................................................................................................................. 3 2.4 Declared Rare and Priority Flora......................................................................................................... 3 2.5 Local and Regional Significance........................................................................................................... 5 2.6 Threatened -

Modelling the Distribution of Photosynthetic Types of Grasses in Sahelian Burkina Faso with High-Resolution Satellite Data
ECOTROPICA 17: 53–63, 2011 © Society for Tropical Ecology MODELLING THE DISTRIBUTION OF PHOTOSYNTHETIC TYPES OF GRASSES IN SAHELIAN BURKINA FASO WITH HIGH-RESOLUTION SATELLITE DATA Marco Schmidt1,2,3*, Konstantin König2,3, Jonas V. Müller4, Ulrike Brunken1,5 & Georg Zizka1,2,3 1 Forschungsinstitut Senckenberg, Abt. Botanik und molekulare Evolutionsforschung, Senckenberganlage 25, 60325 Frankfurt am Main, Germany 2 Goethe-Universität, Institut für Ökologie, Evolution und Diversität, Siesmayerstr. 70, 60323 Frankfurt am Main, Germany 3 Biodiversity and Climate Research Centre (Bik-F), Biodiversity dynamics and Climate, Georg-Voigt-Straße 14-16, 60325 Frankfurt am Main, Germany 4 Royal Botanic Gardens Kew, Seed Conservation Department, Wakehurst Place, Ardingly RH176TN, United Kingdom 5 Palmengarten, Abt. Garten, Wissenschaft & Pädagogik, Siesmayerstr. 61, 60323 Frankfurt am Main, Germany Abstract. We combined grass (Poaceae) occurrence data from the Sahelian parts of Burkina Faso, West Africa, with data on the photosynthetic type of these species. Occurrence data were compiled from relevés and collections of the Herbarium Senckenbergianum, and the assignment of photosynthetic types was taken from the literature and completed by leaf ana- tomical observations of our own. We used the occurrence data to model species distributions using GARP (Genetic algo- rithm of rule-set production) and high-resolution satellite data (Landsat ETM+) as environmental predictors. In a subse- quent step we summarized the distributions of single species for each photosynthetic type. The resulting distribution patterns reflect the ecological preferences connected with photosynthetic pathways. The only C3 species is strictly bound to watercourses and temporary lakes, C4 MS species mainly occur on the dunes, C4 PS-PCK species are mainly from dunes and watercourses, C4 PS-NAD type species dominate the drier peneplains. -
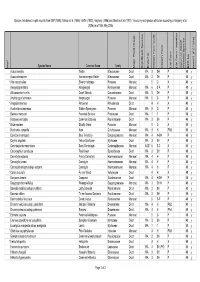
BFS048 Site Species List
Species lists based on plot records from DEP (1996), Gibson et al. (1994), Griffin (1993), Keighery (1996) and Weston et al. (1992). Taxonomy and species attributes according to Keighery et al. (2006) as of 16th May 2005. Species Name Common Name Family Major Plant Group Significant Species Endemic Growth Form Code Growth Form Life Form Life Form - aquatics Common SSCP Wetland Species BFS No kens01 (FCT23a) Wd? Acacia sessilis Wattle Mimosaceae Dicot WA 3 SH P 48 y Acacia stenoptera Narrow-winged Wattle Mimosaceae Dicot WA 3 SH P 48 y * Aira caryophyllea Silvery Hairgrass Poaceae Monocot 5 G A 48 y Alexgeorgea nitens Alexgeorgea Restionaceae Monocot WA 6 S-R P 48 y Allocasuarina humilis Dwarf Sheoak Casuarinaceae Dicot WA 3 SH P 48 y Amphipogon turbinatus Amphipogon Poaceae Monocot WA 5 G P 48 y * Anagallis arvensis Pimpernel Primulaceae Dicot 4 H A 48 y Austrostipa compressa Golden Speargrass Poaceae Monocot WA 5 G P 48 y Banksia menziesii Firewood Banksia Proteaceae Dicot WA 1 T P 48 y Bossiaea eriocarpa Common Bossiaea Papilionaceae Dicot WA 3 SH P 48 y * Briza maxima Blowfly Grass Poaceae Monocot 5 G A 48 y Burchardia congesta Kara Colchicaceae Monocot WA 4 H PAB 48 y Calectasia narragara Blue Tinsel Lily Dasypogonaceae Monocot WA 4 H-SH P 48 y Calytrix angulata Yellow Starflower Myrtaceae Dicot WA 3 SH P 48 y Centrolepis drummondiana Sand Centrolepis Centrolepidaceae Monocot AUST 6 S-C A 48 y Conostephium pendulum Pearlflower Epacridaceae Dicot WA 3 SH P 48 y Conostylis aculeata Prickly Conostylis Haemodoraceae Monocot WA 4 H P 48 y Conostylis juncea Conostylis Haemodoraceae Monocot WA 4 H P 48 y Conostylis setigera subsp. -

Genes Encoding Plastid Acetyl-Coa Carboxylase and 3-Phosphoglycerate Kinase of the Triticum͞aegilops Complex and the Evolutionary History of Polyploid Wheat
Genes encoding plastid acetyl-CoA carboxylase and 3-phosphoglycerate kinase of the Triticum͞Aegilops complex and the evolutionary history of polyploid wheat Shaoxing Huang*†, Anchalee Sirikhachornkit*, Xiujuan Su*, Justin Faris‡§, Bikram Gill‡, Robert Haselkorn*, and Piotr Gornicki*¶ *Department of Molecular Genetics and Cell Biology, University of Chicago, Chicago, IL 60637; and ‡Department of Plant Pathology, Kansas State University, Manhattan, KS 66506 Contributed by Robert Haselkorn, April 12, 2002 The classic wheat evolutionary history is one of adaptive radiation of at the diploid level from a common diploid ancestor and conver- the diploid Triticum͞Aegilops species (A, S, D), genome convergence gence at the polyploid level involving the diverged diploid genomes and divergence of the tetraploid (Triticum turgidum AABB, and (2). Various Aegilops species contributed significantly to the genetic Triticum timopheevii AAGG) and hexaploid (Triticum aestivum, makeup of the polyploid wheats. Extensive classical analyses and AABBDD) species. We analyzed Acc-1 (plastid acetyl-CoA carboxylase) more recent molecular studies have provided information on the and Pgk-1 (plastid 3-phosphoglycerate kinase) genes to determine identity of donors and some of the patterns of genome evolution of phylogenetic relationships among Triticum and Aegilops species of the Triticum͞Aegilops species (3). A review of Triticum and Aegilops the wheat lineage and to establish the timeline of wheat evolution taxonomy and related literature is available from the Wheat based on gene sequence comparisons. Triticum urartu was confirmed Genetic Resource Center at ksu.edu͞wgrc. as the A genome donor of tetraploid and hexaploid wheat. The A The Triticum and Aegilops genera contain 13 diploid and 18 genome of polyploid wheat diverged from T. -

This Article Is from the March 2013 Issue Of
This article is from the March 2013 issue of published by The American Phytopathological Society For more information on this and other topics related to plant pathology, we invite you to visit APSnet at www.apsnet.org Identifying New Sources of Resistance to Eyespot of Wheat in Aegilops longissima H. Sheng and T. D. Murray, Department of Plant Pathology, Washington State University, Pullman 99164-6430 Abstract Sheng, H., and Murray, T. D. 2013. Identifying new sources of resistance to eyespot of wheat in Aegilops longissima. Plant Dis. 97:346-353. Eyespot, caused by Oculimacula yallundae and O. acuformis, is an substitution lines containing chromosomes 1Sl, 2Sl, 5Sl, and 7Sl, and a economically important disease of wheat. Currently, two eyespot re- 4Sl7Sl translocation were resistant to O. yallundae. Chromosomes 1Sl, sistance genes, Pch1 and Pch2, are used in wheat breeding programs 2Sl, 4Sl, and 5Sl contributed to resistance to O. acuformis more than but neither provides complete control or prevents yield loss. Aegilops others. Chromosomes 1Sl, 2Sl, 5Sl, and 7Sl provided resistance to both longissima is a distant relative of wheat and proven donor of genes pathogens. This is the first report of eyespot resistance in A. longis- useful for wheat improvement, including disease resistance. Forty A. sima. These results provide evidence that genetic control of eyespot longissima accessions and 83 A. longissima chromosome addition or resistance is present on multiple chromosomes of the Sl genome. This substitution lines were evaluated for resistance to eyespot. Among the research demonstrates that A. longissima is a potential new source of 40 accessions tested, 43% were resistant to O. -

Bamboo: a Versatile Plant the Journal of the Greens and Gardens, Volume 01, No
An International Journal of Floriculture Science and Landscaping Bamboo: a versatile plant The Journal of the Greens and Gardens, Volume 01, No. 02 : 24-26. 2019 Short Communication Bamboo: a versatile plant Anurag Bajpay* and Kulveer Singh Yadav School of Agricultural Sciences and Technology, Regional Institute of Management and Technology (RIMT) University, Mandi Gobindgarh, Punjab-147301, INDIA *Corresponding Author: [email protected] Received: March 12, 2019 Accepted: March 30, 2019 Abstract This paper deals with the versatile uses of bamboo plant species. It has many useful properties and used for various purposes. Bamboos are used in landscaping, fencing, housing, raw materials for crafts, pulp for paper industry and fabrics for cloth industry besides culinary products, fodder etc. The young shoots of bamboo are used for preparing various delicious foods, processed products and also used as medicines. Various bamboo species are also utilized in herbal and traditional medication. It also has huge potential for solving many problems of environmental and social needs. Key words: Bamboo, environment, culinary, medicine and ornamental use. Introduction: pulp for the paper industry, storage and transportation Bamboo (Bamboo Schreb.) is an evergreen perennial baskets, used in flooring and roofing, preparation of fabrics flowering plant belonging to the monocots family Poaceae. etc. Young rhizomes of Bamboo are widely used to preparing This woody grass widely grows in tropical, subtropical and many kinds of food products. The products of Bamboos are also in temperate zones of India and world as well. The genus gaining popularity among the people and bamboo industries is mainly in distributed Asia, Africa, and Tropical America. -

Survey of Wild Food Plants for Human Consumption in Geçitli (Hakkari, Turkey)
Indian Journal of Traditional Knowledge Vol. 14(2), April 2015, pp. 183-190 Survey of wild food plants for human consumption in Geçitli (Hakkari, Turkey) İdris Kaval1, Lütfi Behçet2 & Uğur Çakilcioğlu3* 1Yuzuncu Yıl University, Department of Biology, Van 65000, Turkey; 2Bingöl University, Department of Biology, Bingöl 12000, Turkey; 3Tunceli University, Pertek Sakine Genç Vocational School, Pertek, Tunceli 62500, Turkey E-mails: [email protected]; [email protected]; [email protected] Received 15 July 2014, revised 22 January 2015 This study aims to record accumulation of knowledge on plants which are used as food by native people of Geçitli (Hakkari, Turkey) that has a rich culture and a very natural environment. In addition, the medical uses of these plants were compiled from the literature. Study area was located on the East of Anatolian diagonal, in the Eastern Anatolia region. Field study was carried out over a period of approximately two years (2008-2010). During this period, 84 vascular plant taxa were collected. The plants were pressed in the field and prepared for identification. A total of 84 food plants belonging to 30 families were identified in the region. In the study being conducted, use of wild plants as food points out interest of people in Geçitli in wild plants. The fact that a large proportion of edible plants are also being used for medicinal purposes indicates that the use of wild plants has a high potential in the region. The present study shows that further ethnobotanical investigations are worthy to be carried out in Turkey, where most of knowledge on popular food plants are still to discover. -

The Complete Plastid Genome Sequence of Iris Gatesii (Section Oncocyclus), a Bearded Species from Southeastern Turkey
Aliso: A Journal of Systematic and Evolutionary Botany Volume 32 | Issue 1 Article 3 2014 The ompletC e Plastid Genome Sequence of Iris gatesii (Section Oncocyclus), a Bearded Species from Southeastern Turkey Carol A. Wilson Rancho Santa Ana Botanic Garden, Claremont, California Follow this and additional works at: http://scholarship.claremont.edu/aliso Part of the Botany Commons, Ecology and Evolutionary Biology Commons, and the Genomics Commons Recommended Citation Wilson, Carol A. (2014) "The ompC lete Plastid Genome Sequence of Iris gatesii (Section Oncocyclus), a Bearded Species from Southeastern Turkey," Aliso: A Journal of Systematic and Evolutionary Botany: Vol. 32: Iss. 1, Article 3. Available at: http://scholarship.claremont.edu/aliso/vol32/iss1/3 Aliso, 32(1), pp. 47–54 ISSN 0065-6275 (print), 2327-2929 (online) THE COMPLETE PLASTID GENOME SEQUENCE OF IRIS GATESII (SECTION ONCOCYCLUS), A BEARDED SPECIES FROM SOUTHEASTERN TURKEY CAROL A. WILSON Rancho Santa Ana Botanic Garden and Claremont Graduate University, 1500 North College Avenue, Claremont, California 91711 ([email protected]) ABSTRACT Iris gatesii is a rare bearded species in subgenus Iris section Oncocyclus that occurs in steppe communities of southeastern Turkey. This species is not commonly cultivated, but related species in section Iris are economically important horticultural plants. The complete plastid genome is reported for I. gatesii based on data generated using the Illumina HiSeq platform and is compared to genomes of 16 species selected from across the monocotyledons. This Iris genome is the only known plastid genome available for order Asparagales that is not from Orchidaceae. The I. gatesii plastid genome, unlike orchid genomes, has little gene loss and rearrangement and is likely to be similar to other genomes from Asparagales. -

Sequence Diversity and Copy Number Variation of Mutator-Like Transposases in Wheat
Genetics and Molecular Biology, 31, 2, 539-546 (2008) Copyright © 2008, Sociedade Brasileira de Genética. Printed in Brazil www.sbg.org.br Research Article Sequence diversity and copy number variation of Mutator-like transposases in wheat Nobuaki Asakura1, Shinya Yoshida2, Naoki Mori3, Ichiro Ohtsuka1 and Chiharu Nakamura3 1Laboratory of Biology, Faculty of Engineering, Kanagawa University, Yokohama, Japan. 2Hyogo Institute of Agriculture, Forestry & Fisheries, Kasai, Japan. 3Laboratory of Plant Genetics, Graduate School of Agricultural Science, Kobe University, Kobe, Japan. Abstract Partial transposase-coding sequences of Mutator-like elements (MULEs) were isolated from a wild einkorn wheat, Triticum urartu, by degenerate PCR. The isolated sequences were classified into a MuDR or Class I clade and di- vided into two distinct subclasses (subclass I and subclass II). The average pair-wise identity between members of both subclasses was 58.8% at the nucleotide sequence level. Sequence diversity of subclass I was larger than that of subclass II. DNA gel blot analysis showed that subclass I was present as low copy number elements in the genomes of all Triticum and Aegilops accessions surveyed, while subclass II was present as high copy number ele- ments. These two subclasses seemed uncapable of recognizing each other for transposition. The number of copies of subclass II elements was much higher in Aegilops with the S, Sl and D genomes and polyploid Triticum species than in diploid Triticum with the A genome, indicating that active transposition occurred in S, Sl and D genomes be- fore polyploidization. DNA gel blot analysis of six species selected from three subfamilies of Poaceae demonstrated that only the tribe Triticeae possessed both subclasses.