In Silico Methods in Prediction of Drug Metabolism, Mass Fragmentation, and Chromatographic Behavior
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -

Prohibited Substances List
Prohibited Substances List This is the Equine Prohibited Substances List that was voted in at the FEI General Assembly in November 2009 alongside the new Equine Anti-Doping and Controlled Medication Regulations(EADCMR). Neither the List nor the EADCM Regulations are in current usage. Both come into effect on 1 January 2010. The current list of FEI prohibited substances remains in effect until 31 December 2009 and can be found at Annex II Vet Regs (11th edition) Changes in this List : Shaded row means that either removed or allowed at certain limits only SUBSTANCE ACTIVITY Banned Substances 1 Acebutolol Beta blocker 2 Acefylline Bronchodilator 3 Acemetacin NSAID 4 Acenocoumarol Anticoagulant 5 Acetanilid Analgesic/anti-pyretic 6 Acetohexamide Pancreatic stimulant 7 Acetominophen (Paracetamol) Analgesic/anti-pyretic 8 Acetophenazine Antipsychotic 9 Acetylmorphine Narcotic 10 Adinazolam Anxiolytic 11 Adiphenine Anti-spasmodic 12 Adrafinil Stimulant 13 Adrenaline Stimulant 14 Adrenochrome Haemostatic 15 Alclofenac NSAID 16 Alcuronium Muscle relaxant 17 Aldosterone Hormone 18 Alfentanil Narcotic 19 Allopurinol Xanthine oxidase inhibitor (anti-hyperuricaemia) 20 Almotriptan 5 HT agonist (anti-migraine) 21 Alphadolone acetate Neurosteriod 22 Alphaprodine Opiod analgesic 23 Alpidem Anxiolytic 24 Alprazolam Anxiolytic 25 Alprenolol Beta blocker 26 Althesin IV anaesthetic 27 Althiazide Diuretic 28 Altrenogest (in males and gelidngs) Oestrus suppression 29 Alverine Antispasmodic 30 Amantadine Dopaminergic 31 Ambenonium Cholinesterase inhibition 32 Ambucetamide Antispasmodic 33 Amethocaine Local anaesthetic 34 Amfepramone Stimulant 35 Amfetaminil Stimulant 36 Amidephrine Vasoconstrictor 37 Amiloride Diuretic 1 Prohibited Substances List This is the Equine Prohibited Substances List that was voted in at the FEI General Assembly in November 2009 alongside the new Equine Anti-Doping and Controlled Medication Regulations(EADCMR). -

Pharmaceuticals Appendix
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ADAPALENE 106685-40-9 ABANOQUIL 90402-40-7 ADAPROLOL 101479-70-3 ABECARNIL 111841-85-1 ADEMETIONINE 17176-17-9 ABLUKAST 96566-25-5 ADENOSINE PHOSPHATE 61-19-8 ABUNIDAZOLE 91017-58-2 ADIBENDAN 100510-33-6 ACADESINE 2627-69-2 ADICILLIN 525-94-0 ACAMPROSATE 77337-76-9 ADIMOLOL 78459-19-5 ACAPRAZINE 55485-20-6 ADINAZOLAM 37115-32-5 ACARBOSE 56180-94-0 ADIPHENINE 64-95-9 ACEBROCHOL 514-50-1 ADIPIODONE 606-17-7 ACEBURIC ACID 26976-72-7 ADITEREN 56066-19-4 ACEBUTOLOL 37517-30-9 ADITOPRIME 56066-63-8 ACECAINIDE 32795-44-1 ADOSOPINE 88124-26-9 ACECARBROMAL 77-66-7 ADOZELESIN 110314-48-2 ACECLIDINE 827-61-2 ADRAFINIL 63547-13-7 ACECLOFENAC 89796-99-6 ADRENALONE 99-45-6 ACEDAPSONE 77-46-3 AFALANINE 2901-75-9 ACEDIASULFONE SODIUM 127-60-6 AFLOQUALONE 56287-74-2 ACEDOBEN 556-08-1 AFUROLOL 65776-67-2 ACEFLURANOL 80595-73-9 AGANODINE 86696-87-9 ACEFURTIAMINE 10072-48-7 AKLOMIDE 3011-89-0 ACEFYLLINE CLOFIBROL 70788-27-1 -
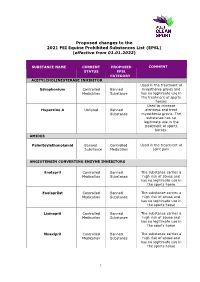
Proposed Changes to the 2021 FEI Equine Prohibited Substances List (EPSL) (Effective from 01.01.2022)
Proposed changes to the 2021 FEI Equine Prohibited Substances List (EPSL) (effective from 01.01.2022) SUBSTANCE NAME CURRENT PROPOSED COMMENT STATUS EPSL CATEGORY ACETYLCHOLINESTERASE INHIBITOR Used in the treatment of Edrophonium Controlled Banned myasthenia gravis and Medication Substance has no legitimate use in the treatment of sports horses Used to increase Huperzine A Unlisted Banned alertness and treat Substance myasthenia gravis. The substance has no legitimate use in the treatment of sports horses. AMIDES Palmitoylethanolamid Banned Controlled Used in the treatment of Substance Medication joint pain ANGIOTENSIN CONVERTING ENZYME INHIBITORS Enalapril Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse Enalaprilat Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse Lisinopril Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse Moexipril Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse 1 Perindoprilat Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse ANTIHISTAMINES Antazoline Controlled Banned The substance has no Medication Substance legitimate use in the sports horse Azatadine Controlled Banned The substance has Medication Substance sedative effects -

BMJ Open Is Committed to Open Peer Review. As Part of This Commitment We Make the Peer Review History of Every Article We Publish Publicly Available
BMJ Open is committed to open peer review. As part of this commitment we make the peer review history of every article we publish publicly available. When an article is published we post the peer reviewers’ comments and the authors’ responses online. We also post the versions of the paper that were used during peer review. These are the versions that the peer review comments apply to. The versions of the paper that follow are the versions that were submitted during the peer review process. They are not the versions of record or the final published versions. They should not be cited or distributed as the published version of this manuscript. BMJ Open is an open access journal and the full, final, typeset and author-corrected version of record of the manuscript is available on our site with no access controls, subscription charges or pay-per-view fees (http://bmjopen.bmj.com). If you have any questions on BMJ Open’s open peer review process please email [email protected] BMJ Open Pediatric drug utilization in the Western Pacific region: Australia, Japan, South Korea, Hong Kong and Taiwan Journal: BMJ Open ManuscriptFor ID peerbmjopen-2019-032426 review only Article Type: Research Date Submitted by the 27-Jun-2019 Author: Complete List of Authors: Brauer, Ruth; University College London, Research Department of Practice and Policy, School of Pharmacy Wong, Ian; University College London, Research Department of Practice and Policy, School of Pharmacy; University of Hong Kong, Centre for Safe Medication Practice and Research, Department -

The Organic Chemistry of Drug Synthesis
THE ORGANIC CHEMISTRY OF DRUG SYNTHESIS VOLUME 3 DANIEL LEDNICER Analytical Bio-Chemistry Laboratories, Inc. Columbia, Missouri LESTER A. MITSCHER The University of Kansas School of Pharmacy Department of Medicinal Chemistry Lawrence, Kansas A WILEY-INTERSCIENCE PUBLICATION JOHN WILEY AND SONS New York • Chlchester • Brisbane * Toronto • Singapore Copyright © 1984 by John Wiley & Sons, Inc. All rights reserved. Published simultaneously in Canada. Reproduction or translation of any part of this work beyond that permitted by Section 107 or 108 of the 1976 United States Copyright Act without the permission of the copyright owner is unlawful. Requests for permission or further information should be addressed to the Permissions Department, John Wiley & Sons, Inc. Library of Congress Cataloging In Publication Data: (Revised for volume 3) Lednicer, Daniel, 1929- The organic chemistry of drug synthesis. "A Wiley-lnterscience publication." Includes bibliographical references and index. 1. Chemistry, Pharmaceutical. 2. Drugs. 3. Chemistry, Organic—Synthesis. I. Mitscher, Lester A., joint author. II. Title. [DNLM 1. Chemistry, Organic. 2. Chemistry, Pharmaceutical. 3. Drugs—Chemical synthesis. QV 744 L473o 1977] RS403.L38 615M9 76-28387 ISBN 0-471-09250-9 (v. 3) Printed in the United States of America 10 907654321 With great pleasure we dedicate this book, too, to our wives, Beryle and Betty. The great tragedy of Science is the slaying of a beautiful hypothesis by an ugly fact. Thomas H. Huxley, "Biogenesis and Abiogenisis" Preface Ihe first volume in this series represented the launching of a trial balloon on the part of the authors. In the first place, wo were not entirely convinced that contemporary medicinal (hemistry could in fact be organized coherently on the basis of organic chemistry. -

(12) Patent Application Publication (10) Pub. No.: US 2006/0078604 A1 Kanios Et Al
US 20060078604A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2006/0078604 A1 Kanios et al. (43) Pub. Date: Apr. 13, 2006 (54) TRANSDERMAL DRUG DELIVERY DEVICE Related U.S. Application Data INCLUDING AN OCCLUSIVE BACKING (60) Provisional application No. 60/616,861, filed on Oct. 8, 2004. (75) Inventors: David Kanios, Miami, FL (US); Juan A. Mantelle, Miami, FL (US); Viet Publication Classification Nguyen, Miami, FL (US) (51) Int. Cl. Correspondence Address: A 6LX 9/70 (2006.01) DCKSTEIN SHAPRO MORN & OSHINSKY (52) U.S. Cl. .............................................................. 424/449 LLP (57) ABSTRACT 2101 L Street, NW Washington, DC 20037 (US) A transdermal drug delivery system for the topical applica tion of one or more active agents contained in one or more (73) Assignee: Noven Pharmaceuticals, Inc. polymeric and/or adhesive carrier layers, proximate to a non-drug containing polymeric backing layer which can (21) Appl. No.: 11/245,180 control the delivery rate and profile of the transdermal drug delivery system by adjusting the moisture vapor transmis (22) Filed: Oct. 7, 2005 sion rate of the polymeric backing layer. Patent Application Publication Apr. 13, 2006 Sheet 1 of 2 US 2006/0078604 A1 Fis ZZZZZZZZZZZZZZZZZZZ :::::::::::::::::::::::::::::::: Patent Application Publication Apr. 13, 2006 Sheet 2 of 2 US 2006/0078604 A1 3. s s 3. a 3 : 8 g US 2006/0078604 A1 Apr. 13, 2006 TRANSIDERMAL DRUG DELVERY DEVICE 0008. In the “classic' reservoir-type device, the active INCLUDING AN OCCLUSIVE BACKING agent is typically dissolved or dispersed in a carrier to yield a non-finite carrier form, Such as, for example, a fluid or gel. -

Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DIX to the HTSUS—Continued
20558 Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DEPARMENT OF THE TREASURY Services, U.S. Customs Service, 1301 TABLE 1.ÐPHARMACEUTICAL APPEN- Constitution Avenue NW, Washington, DIX TO THE HTSUSÐContinued Customs Service D.C. 20229 at (202) 927±1060. CAS No. Pharmaceutical [T.D. 95±33] Dated: April 14, 1995. 52±78±8 ..................... NORETHANDROLONE. A. W. Tennant, 52±86±8 ..................... HALOPERIDOL. Pharmaceutical Tables 1 and 3 of the Director, Office of Laboratories and Scientific 52±88±0 ..................... ATROPINE METHONITRATE. HTSUS 52±90±4 ..................... CYSTEINE. Services. 53±03±2 ..................... PREDNISONE. 53±06±5 ..................... CORTISONE. AGENCY: Customs Service, Department TABLE 1.ÐPHARMACEUTICAL 53±10±1 ..................... HYDROXYDIONE SODIUM SUCCI- of the Treasury. NATE. APPENDIX TO THE HTSUS 53±16±7 ..................... ESTRONE. ACTION: Listing of the products found in 53±18±9 ..................... BIETASERPINE. Table 1 and Table 3 of the CAS No. Pharmaceutical 53±19±0 ..................... MITOTANE. 53±31±6 ..................... MEDIBAZINE. Pharmaceutical Appendix to the N/A ............................. ACTAGARDIN. 53±33±8 ..................... PARAMETHASONE. Harmonized Tariff Schedule of the N/A ............................. ARDACIN. 53±34±9 ..................... FLUPREDNISOLONE. N/A ............................. BICIROMAB. 53±39±4 ..................... OXANDROLONE. United States of America in Chemical N/A ............................. CELUCLORAL. 53±43±0 -

2 12/ 35 74Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 22 March 2012 (22.03.2012) 2 12/ 35 74 Al (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 9/16 (2006.01) A61K 9/51 (2006.01) kind of national protection available): AE, AG, AL, AM, A61K 9/14 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, (21) International Application Number: DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, PCT/EP201 1/065959 HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, (22) International Filing Date: KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, 14 September 201 1 (14.09.201 1) ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, (25) Filing Language: English RW, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, (26) Publication Language: English TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 61/382,653 14 September 2010 (14.09.2010) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant (for all designated States except US): GM, KE, LR, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, NANOLOGICA AB [SE/SE]; P.O Box 8182, S-104 20 ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, Stockholm (SE). -

Drugs, Poisons and Controlled Substances Act 1981
1834 ANNO TRICESIMO ELIZABETHAE SECUNDAE REGINAE VICTORIA Drugs, Poisons and Controlled Substances Act 1981 No. 9719 An Act to re-enact with Amendments the Law relating to Drugs, Poisons and Controlled Substances, to amend the Health Act 1958 and the Crimes Act 1958 and for other purposes. [Assented to 12 January 1982] BE IT ENACTED by the Queen's Most Excellent Majesty by and with the advice and consent of the Legislative Council and the Legislative Assembly of Victoria in this present ParUament assembled and by the authority of the same as follows (that is to say): Short tiUe. 1. (1) This Act may be cited as the Drugs, Poisons and Controlled Substances y4cM981. Commenco- (2) The several provisions of this Act shall come into operation ment. on a day or on the respective days to be fixed by proclamation or successive proclamations of the Governor in Council published in the Government Gazette. Arrangement (3) This Act is divided into Parts and Divisions as follows: of Act. Part I.—Introductory and Transitional ss. 2-11. Part II.—Poisons and Controlled Substances ss. 12-55. Division 1—Classification s. 12. Division 2—Authorized Persons ss. 13-14. Division 1981 Drugs, Poisons and Controlled Substances No. 9719 2335 Division 3—Poisons Advisory Committee ss. 15-18. Division 4—Licensed Persons s. 19. Division 5—Special Poisons s. 20. Division 6—Industrial, Educational and Laboratory Permits s. 21. Division 7—Issue of Licences and Permits s. 22. Division 8—Manufacture and Sale of Poisons or . Controlled Substances ss. 23-30. Division 9—Trade or Proprietary Poisons s. -

Final1-Aritmo-Final-Report-V2-0Final.Pdf
ARITMO Final Report PROJECT FINAL REPORT Grant Agreement number: 241679 Project acronym: ARITMO Project title: Arrhythmogenic potential of drugs Funding Scheme: Small or Medium-Scale Focused Research Project Period covered: from 1st January 2010 to 30th June 2013 Name of the scientific representative of the project's co-ordinator, Title and Organisation: Prof. Miriam CJM Sturkenboom, Erasmus Universitair Medisch Centrum Rotterdam Tel: +31 10 704 4126 Fax: +31 10 704 4722 E-mail: [email protected] Project website1 address: www.aritmo-project.org 1 The home page of the website should contain the generic European flag and the FP7 logo which are available in electronic format at the Europa website (logo of the European flag: http://europa.eu/abc/symbols/emblem/index_en.htm ; logo of the 7th FP: http://ec.europa.eu/research/fp7/index_en.cfm?pg=logos). The area of activity of the project should also be mentioned. © Copyright 2013 ARITMO Consortium 1 ARITMO Final Report Table of contents Table of contents ................................................................................................................................................................. 2 1. Final publishable summary report ................................................................................................................................ 3 1.1 Executive summary ................................................................................................................................................. 3 1.2 Description of project context and -

Common Study Protocol for Observational Database Studies WP5 – Analytic Database Studies
Arrhythmogenic potential of drugs FP7-HEALTH-241679 http://www.aritmo-project.org/ Common Study Protocol for Observational Database Studies WP5 – Analytic Database Studies V 1.3 Draft Lead beneficiary: EMC Date: 03/01/2010 Nature: Report Dissemination level: D5.2 Report on Common Study Protocol for Observational Database Studies WP5: Conduct of Additional Observational Security: Studies. Author(s): Gianluca Trifiro’ (EMC), Giampiero Version: v1.1– 2/85 Mazzaglia (F-SIMG) Draft TABLE OF CONTENTS DOCUMENT INFOOMATION AND HISTORY ...........................................................................4 DEFINITIONS .................................................... ERRORE. IL SEGNALIBRO NON È DEFINITO. ABBREVIATIONS ......................................................................................................................6 1. BACKGROUND .................................................................................................................7 2. STUDY OBJECTIVES................................ ERRORE. IL SEGNALIBRO NON È DEFINITO. 3. METHODS ..........................................................................................................................8 3.1.STUDY DESIGN ....................................................................................................................8 3.2.DATA SOURCES ..................................................................................................................9 3.2.1. IPCI Database .....................................................................................................9