View of the Pichia Expression System
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dual Role of the Mitochondrial Protein Frataxin in Astrocytic Tumors
Laboratory Investigation (2011) 91, 1766–1776 & 2011 USCAP, Inc All rights reserved 0023-6837/11 $32.00 Dual role of the mitochondrial protein frataxin in astrocytic tumors Elmar Kirches1, Nadine Andrae1, Aline Hoefer2, Barbara Kehler1,3, Kim Zarse4, Martin Leverkus3, Gerburg Keilhoff5, Peter Schonfeld5, Thomas Schneider6, Annette Wilisch-Neumann1 and Christian Mawrin1 The mitochondrial protein frataxin (FXN) is known to be involved in mitochondrial iron homeostasis and iron–sulfur cluster biogenesis. It is discussed to modulate function of the electron transport chain and production of reactive oxygen species (ROS). FXN loss in neurons and heart muscle cells causes an autosomal-dominant mitochondrial disorder, Friedreich’s ataxia. Recently, tumor induction after targeted FXN deletion in liver and reversal of the tumorigenic phenotype of colonic carcinoma cells following FXN overexpression were described in the literature, suggesting a tumor suppressor function. We hypothesized that a partial reversal of the malignant phenotype of glioma cells should occur after FXN transfection, if the mitochondrial protein has tumor suppressor functions in these brain tumors. In astrocytic brain tumors and tumor cell lines, we observed reduced FXN levels compared with non-neoplastic astrocytes. Mitochondrial content (citrate synthase activity) was not significantly altered in U87MG glioblastoma cells stably overexpressing FXN (U87-FXN). Surprisingly, U87-FXN cells exhibited increased cytoplasmic ROS levels, although mitochondrial ROS release was attenuated by FXN, as expected. Higher cytoplasmic ROS levels corresponded to reduced activities of glutathione peroxidase and catalase, and lower glutathione content. The defect of antioxidative capacity resulted in increased susceptibility of U87-FXN cells against oxidative stress induced by H2O2 or buthionine sulfoximine. -

ARTICLES Functional Mitochondrial Heterogeneity in Heteroplasmic
0031-3998/00/4802-0143 PEDIATRIC RESEARCH Vol. 48, No. 2, 2000 Copyright © 2000 International Pediatric Research Foundation, Inc. Printed in U.S.A. ARTICLES Functional Mitochondrial Heterogeneity in Heteroplasmic Cells Carrying the Mitochondrial DNA Mutation Associated with the MELAS Syndrome (Mitochondrial Encephalopathy, Lactic Acidosis, and Strokelike Episodes) ANNETTE BAKKER, CYRILLE BARTHE´ LE´ MY, PAULE FRACHON, DANIELLE CHATEAU, DAMIEN STERNBERG, JEAN PIERRE MAZAT, AND ANNE LOMBE` S INSERM UR523, Institut de Myologie, 75651 Paris, France [A.B., C.B., P.F., D.C., A.L.]; Biochimie B, Hoˆpital de La Salpeˆtrie`re, 75651 Paris, France [D.S.]; and Universite´ de Bordeaux II, INSERM E99–29, 33076 Bordeaux cedex, France [J.P.M.] ABSTRACT Most mitochondrial DNA (mtDNA) alterations associated wild-type mtDNA, transfer RNA, or protein. Mitochondria in with human disorders are heteroplasmic, i.e. mutant mtDNA these heteroplasmic cells cannot, therefore, be considered a molecules coexist with normal ones within the cell. We ad- single functional unit. (Pediatr Res 48: 143–150, 2000) dressed the possibility of intermitochondrial exchanges through histologic analyses of cybrid clones with increasing proportion of the MELAS (A3243G) mtDNA transfer RNA point mutation. Abbreviations MtDNA-dependent cytochrome c oxidase activity and protein MELAS, mitochondrial myopathy, encephalopathy, lactic composition as well as mitochondrial membrane potential ap- acidosis, and strokelike episodes peared heterogeneous in individual cells from clonal heteroplas- -

Stouthamer1973
Antonie van Leeuwenhoek 39 (1973) 545-565 545 A theoretical study on the amount of ATP required for synthesis of microbial cell material A. H. STOUTHAMER Biological Laboratory, Free University, de Boelelaan 1087, Amsterdam, the Netherlands STOUTHAMER, A.H. 1973. A theoretical study on the amount of ATP required for synthesis of microbial cell material. Antonie van Leeuwenhoek 39: 545-565. The amount of ATP required for the formation of microbial cells growing under various conditions was calculated. It was assumed that the chemical com position of the cell was the same under all these conditions. The analysis of the chemical composition of microbial cells of Morowitz ( 1968) was taken as a base. It was assumed that 4 moles of ATP are required for the incorporation of one mole of amino acid into protein. The amount of ATP required on account of the instability and frequent regeneration of messenger RNA was calculated from data in the literature pertaining to the relative rates of synthesis of the various classes of RNA molecules in the cell. An estimate is given of the amount of ATP required for transport processes. For this purpose it was assumed that 0.5 mole of ATP is necessary for the uptake of 1 g-ion of potassium or ammo nium, and 1 mole of ATP for the uptake of 1 mole of phosphate, amino acid, acetate, malate etc. The results of the calculations show that from preformed monomers (glucose, amino acids and nucleic acid bases) 31.9 g cells can be formed per g-mole of ATP when acetyl-CoA is formed from glucose. -

The Pathogenetic Role of Β-Cell Mitochondria in Type 2 Diabetes
236 3 Journal of M Fex et al. Mitochondria in β-cells 236:3 R145–R159 Endocrinology REVIEW The pathogenetic role of β-cell mitochondria in type 2 diabetes Malin Fex1, Lisa M Nicholas1, Neelanjan Vishnu1, Anya Medina1, Vladimir V Sharoyko1, David G Nicholls1, Peter Spégel1,2 and Hindrik Mulder1 1Department of Clinical Sciences in Malmö, Unit of Molecular Metabolism, Lund University Diabetes Centre, Clinical Research Center, Malmö University Hospital, Lund University, Malmö, Sweden 2Department of Chemistry, Center for Analysis and Synthesis, Lund University, Sweden Correspondence should be addressed to H Mulder: [email protected] Abstract Mitochondrial metabolism is a major determinant of insulin secretion from pancreatic Key Words β-cells. Type 2 diabetes evolves when β-cells fail to release appropriate amounts f TCA cycle of insulin in response to glucose. This results in hyperglycemia and metabolic f coupling signal dysregulation. Evidence has recently been mounting that mitochondrial dysfunction f oxidative phosphorylation plays an important role in these processes. Monogenic dysfunction of mitochondria is a f mitochondrial transcription rare condition but causes a type 2 diabetes-like syndrome owing to β-cell failure. Here, f genetic variation we describe novel advances in research on mitochondrial dysfunction in the β-cell in type 2 diabetes, with a focus on human studies. Relevant studies in animal and cell models of the disease are described. Transcriptional and translational regulation in mitochondria are particularly emphasized. The role of metabolic enzymes and pathways and their impact on β-cell function in type 2 diabetes pathophysiology are discussed. The role of genetic variation in mitochondrial function leading to type 2 diabetes is highlighted. -

Progressive Increase in Mtdna 3243A>G Heteroplasmy Causes
Progressive increase in mtDNA 3243A>G PNAS PLUS heteroplasmy causes abrupt transcriptional reprogramming Martin Picarda, Jiangwen Zhangb, Saege Hancockc, Olga Derbenevaa, Ryan Golhard, Pawel Golike, Sean O’Hearnf, Shawn Levyg, Prasanth Potluria, Maria Lvovaa, Antonio Davilaa, Chun Shi Lina, Juan Carlos Perinh, Eric F. Rappaporth, Hakon Hakonarsonc, Ian A. Trouncei, Vincent Procaccioj, and Douglas C. Wallacea,1 aCenter for Mitochondrial and Epigenomic Medicine, Children’s Hospital of Philadelphia and the Department of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, PA 19104; bSchool of Biological Sciences, The University of Hong Kong, Hong Kong, People’s Republic of China; cTrovagene, San Diego, CA 92130; dCenter for Applied Genomics, Division of Genetics, Department of Pediatrics, and hNucleic Acid/Protein Research Core Facility, Children’s Hospital of Philadelphia, Philadelphia, PA 19104; eInstitute of Genetics and Biotechnology, Warsaw University, 00-927, Warsaw, Poland; fMorton Mower Central Research Laboratory, Sinai Hospital of Baltimore, Baltimore, MD 21215; gGenomics Sevices Laboratory, HudsonAlpha Institute for Biotechnology, Huntsville, AL 35806; iCentre for Eye Research Australia, Royal Victorian Eye and Ear Hospital, East Melbourne, VIC 3002, Australia; and jDepartment of Biochemistry and Genetics, National Center for Neurodegenerative and Mitochondrial Diseases, Centre Hospitalier Universitaire d’Angers, 49933 Angers, France Contributed by Douglas C. Wallace, August 1, 2014 (sent for review May -

Src Inhibitors Modulate Frataxin Protein Levels
Human Molecular Genetics, 2015, Vol. 24, No. 15 4296–4305 doi: 10.1093/hmg/ddv162 Advance Access Publication Date: 6 May 2015 Original Article ORIGINAL ARTICLE Src inhibitors modulate frataxin protein levels Downloaded from https://academic.oup.com/hmg/article/24/15/4296/2453025 by guest on 28 September 2021 Fabio Cherubini1, Dario Serio1, Ilaria Guccini1, Silvia Fortuni1,2, Gaetano Arcuri1, Ivano Condò1, Alessandra Rufini1,2, Shadman Moiz1, Serena Camerini3, Marco Crescenzi3, Roberto Testi1,2 and Florence Malisan1,* 1Laboratory of Signal Transduction, Department of Biomedicine and Prevention, University of Rome ‘Tor Vergata’, Via Montpellier 1, 00133 Rome, Italy, 2Fratagene Therapeutics Ltd, 22 Northumberland Rd, Dublin, Ireland and 3Department of Cell Biology and Neurosciences, Italian National Institute of Health, Viale Regina Elena, 299, 00161 Rome, Italy *To whom correspondence should be addressed at: Laboratory of Signal Transduction, Department of Biomedicine and Prevention, University of Rome ‘Tor Vergata’, Via Montpellier 1, 00133 Rome, Italy. Tel: +39 0672596501; Fax: +39 0672596505; Email: [email protected] Abstract Defective expression of frataxin is responsible for the inherited, progressive degenerative disease Friedreich’s Ataxia (FRDA). There is currently no effective approved treatment for FRDA and patients die prematurely. Defective frataxin expression causes critical metabolic changes, including redox imbalance and ATP deficiency. As these alterations are known to regulate the tyrosine kinase Src, we investigated whether Src might in turn affect frataxin expression. We found that frataxin can be phosphorylated by Src. Phosphorylation occurs primarily on Y118 and promotes frataxin ubiquitination, a signal for degradation. Accordingly, Src inhibitors induce accumulation of frataxin but are ineffective on a non-phosphorylatable frataxin- Y118F mutant. -

Characterization of Two Cdnas Encoding Mitochondrial Lipoamide Dehydrogenase from Arabidopsis Isabelle Lutziger Iowa State University
Botany Publication and Papers Botany 10-2001 Characterization of Two cDNAs Encoding Mitochondrial Lipoamide Dehydrogenase from Arabidopsis Isabelle Lutziger Iowa State University David J. Oliver Iowa State University, [email protected] Follow this and additional works at: http://lib.dr.iastate.edu/bot_pubs Part of the Botany Commons Recommended Citation Lutziger, Isabelle and Oliver, David J., "Characterization of Two cDNAs Encoding Mitochondrial Lipoamide Dehydrogenase from Arabidopsis" (2001). Botany Publication and Papers. 1. http://lib.dr.iastate.edu/bot_pubs/1 This Article is brought to you for free and open access by the Botany at Iowa State University Digital Repository. It has been accepted for inclusion in Botany Publication and Papers by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Characterization of Two cDNAs Encoding Mitochondrial Lipoamide Dehydrogenase from Arabidopsis Abstract In contrast to peas (Pisum sativum), where mitochondrial lipoamide dehydrogenase is encoded by a single gene and shared between the α-ketoacid dehydrogenase complexes and the Gly decarboxylase complex, Arabidopsis has two genes encoding for two mitochondrial lipoamide dehydrogenases. Northern-blot analysis revealed different levels of RNA expression for the two genes in different organs; mtLPD1 had higher RNA levels in green leaves compared with the much lower level in roots. The mRNA formtLPD2 shows the inverse pattern. The other organs examined showed nearly equal RNA expressions for both genes. Analysis of etiolated seedlings transferred to light showed a strong induction of RNA expression for mtLPD1 but only a moderate induction of mtLPD2. Based on the organ and light-dependent expression patterns, we hypothesize thatmtLPD1encodes the protein most often associated with the Gly decarboxylase complex, and mtLPD2 encodes the protein incorporated into α-ketoacid dehydrogenase complexes. -

Shared Sulfur Mobilization Routes for Trna Thiolation and Molybdenum Cofactor Biosynthesis in Prokaryotes and Eukaryotes
biomolecules Review Shared Sulfur Mobilization Routes for tRNA Thiolation and Molybdenum Cofactor Biosynthesis in Prokaryotes and Eukaryotes Silke Leimkühler *, Martin Bühning and Lena Beilschmidt Department of Molecular Enzymology, Institute of Biochemistry and Biology, University of Potsdam, 14476 Potsdam, Germany; [email protected] (M.B.); [email protected] (L.B.) * Correspondence: [email protected]; Tel.: +49-331-977-5603 Academic Editor: Valérie de Crécy-Lagard Received: 8 December 2016; Accepted: 9 January 2017; Published: 14 January 2017 Abstract: Modifications of transfer RNA (tRNA) have been shown to play critical roles in the biogenesis, metabolism, structural stability and function of RNA molecules, and the specific modifications of nucleobases with sulfur atoms in tRNA are present in pro- and eukaryotes. Here, especially the thiomodifications xm5s2U at the wobble position 34 in tRNAs for Lys, Gln and Glu, were suggested to have an important role during the translation process by ensuring accurate deciphering of the genetic code and by stabilization of the tRNA structure. The trafficking and delivery of sulfur nucleosides is a complex process carried out by sulfur relay systems involving numerous proteins, which not only deliver sulfur to the specific tRNAs but also to other sulfur-containing molecules including iron–sulfur clusters, thiamin, biotin, lipoic acid and molybdopterin (MPT). Among the biosynthesis of these sulfur-containing molecules, the biosynthesis of the molybdenum cofactor (Moco) and the synthesis of thio-modified tRNAs in particular show a surprising link by sharing protein components for sulfur mobilization in pro- and eukaryotes. Keywords: tRNA; molybdenum cofactor; persulfide; thiocarboxylate; thionucleosides; sulfurtransferase; L-cysteine desulfurase 1. -

Molecular Basis of Dihydrouridine Formation on Trna
Molecular basis of dihydrouridine formation on tRNA Futao Yua1, Yoshikazu Tanakab,c1, Keitaro Yamashitaa, Takeo Suzukid, Akiyoshi Nakamurac, Nagisa Hiranoc, Tsutomu Suzukid, Min Yaoa,c, and Isao Tanakaa,c,2 aGraduate School of Life Sciences, Hokkaido University, Sapporo 060-0810, Japan; bCreative Research Institution “Sousei,” Hokkaido University, Sapporo 001-0021, Japan; cFaculty of Advanced Life Sciences, Hokkaido University, Sapporo 060-0810, Japan; and dDepartment of Chemistry and Biotechnology, School of Engineering, University of Tokyo, Tokyo 113-8656, Japan Edited by Dieter Söll, Yale University, New Haven, CT, and approved October 3, 2011 (received for review July 28, 2011) Dihydrouridine (D) is a highly conserved modified base found Similarly, the site specificity and nonredundant catalytic func- in tRNAs from all domains of life. Dihydrouridine synthase (Dus) tions were also confirmed in three Dus from E. coli (YjbN, YhdG, catalyzes the D formation of tRNA through reduction of uracil base and YohI) (8). The crystal structure of Dus from Thermotoga with flavin mononucleotide (FMN) as a cofactor. Here, we report maritima has been reported (9), and mutation analysis of Dus the crystal structures of Thermus thermophilus Dus (TthDus), which from E. coli revealed important residues for dihydrouridine is responsible for D formation at positions 20 and 20a, in complex formation (10). One of the most remarkable biochemical features with tRNA and with a short fragment of tRNA (D-loop). Dus inter- of Dus is that other modifications of tRNA are required for the acts extensively with the D-arm and recognizes the elbow region enzymatic activity (11). However, the details of the reaction, composed of the kissing loop interaction between T- and D-loops in including the tRNA recognition mechanism and its catalysis of tRNA, pulling U20 into the catalytic center for reduction. -

Rrna and Trna Bridges to Neuronal Homeostasis in Health and Disease
Review rRNA and tRNA Bridges to Neuronal Homeostasis in Health and Disease Francesca Tuorto 1 and Rosanna Parlato 2,3 1 - Division of Epigenetics, DKFZ-ZMBH Alliance, German Cancer Research Center, Im Neuenheimer Feld 580, 69120 Heidelberg, Germany 2 - Institute of Applied Physiology, University of Ulm, Albert Einstein Allee 11, 89081 Ulm, Germany 3 - Institute of Anatomy and Cell Biology, Medical Cell Biology, University of Heidelberg, Im Neuenheimer Feld 307, 69120 Heidelberg, Germany Correspondence to Francesca Tuorto and Rosanna Parlato: German Cancer Research Center, Im Neuenheimer Feld 580, 69120 Heidelberg, Germany. University of Ulm, Institute of Applied Physiology, Albert Einstein Allee, 11, 89081 Ulm, Germany. [email protected], [email protected] https://doi.org/10.1016/j.jmb.2019.03.004 Edited by Ernst Stefan Seemann Abstract Dysregulation of protein translation is emerging as a unifying mechanism in the pathogenesis of many neuronal disorders. Ribosomal RNA (rRNA) and transfer RNA (tRNA) are structural molecules that have complementary and coordinated functions in protein synthesis. Defects in both rRNAs and tRNAs have been described in mammalian brain development, neurological syndromes, and neurodegeneration. In this review, we present the molecular mechanisms that link aberrant rRNA and tRNA transcription, processing and modifications to translation deficits, and neuropathogenesis. We also discuss the interdependence of rRNA and tRNA biosynthesis and how their metabolism brings together proteotoxic stress and impaired neuronal homeostasis. © 2019 Elsevier Ltd. All rights reserved. Introduction at specific positions [4]. There are two main types of tsRNAs: tRNA-derived fragments (tRFs; 14–30 Types of RNA in a eukaryotic cell classically nucleotides) and tiRNAs (tRNA-derived stress- included ribosomal RNA (rRNA), transfer RNA induced RNAs, or tRNA halves, 28–36 nucleotides), (tRNA), and messenger RNA (mRNA). -
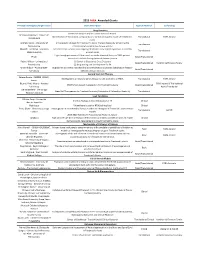
Grantsummaries 2014-2017 Jf
2015 FARA Awarded Grants Principal Investigator/Organization Grant Description Type of Research Co-funding Drug Discovery 2014 Keith Michael Andrus Cardiac Research Award: Veronique Monnier - Université Identification of therapeutic compounds on a cardiac Drosophila model of Friedreich’s Translational FARA Ireland Paris Diderot ataxia Andrew Dancis - University of A therapeutic strategy for Friedreich's ataxia: Frataxin bypass by enhancing the Translational Pennsylvania mitochondrial cysteine desulfurase activity Edward L. Grabczyk - Louisiana Small molecule induced exon skipping of MLH3 to slow repeat expansion in an FRDA Translational State University mousel model High throughput screen of Pfizer small molecule chemical library in FRDA patient- Pfizer Basic/Translational derived cells to look for regulators of frataxin protein Robert Wilson - University of (1) Center of Excellence: Drug Discovery Basic/Translational Hamilton & Finneran Family Pennsylvania (2) Drug testing and development for FA Omar Khdour - Arizona State Approaches to correct mitochondrial abnormalities and synaptic pathology in frataxin- Basic/Translational University deficient sensory neurons Gene & Stem Cell Therapy Helene Puccio - INSERM, IGBMC, Development of neuronal gene therapy for the treatment of FRDA Translational FARA Ireland France Ricardo Pinto Mouro - Harvard FARA Ireland & The National CRISPR/Cas9-based Therapeutics for Friedreich's ataxia Basic/Translational University Ataxia Foundation Joel Gottesfeld - The Scripps Stem Cell Therapeutics for Friedreich's -

Oxidative Stress, Mitochondrial Dysfunction, and Neuroprotection of Polyphenols with Respect to Resveratrol in Parkinson’S Disease
biomedicines Review Oxidative Stress, Mitochondrial Dysfunction, and Neuroprotection of Polyphenols with Respect to Resveratrol in Parkinson’s Disease Heng-Chung Kung 1,2 , Kai-Jung Lin 1,3 , Chia-Te Kung 4,* and Tsu-Kung Lin 1,5,6,* 1 Center for Mitochondrial Research and Medicine, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83301, Taiwan; [email protected] (H.-C.K.); [email protected] (K.-J.L.) 2 Department of Biology, Krieger School of Arts and Sciences, Johns Hopkins University, Baltimore, MD 21218, USA 3 Department of Family Medicine, National Taiwan University Hospital, Taipei 100225, Taiwan 4 Department of Emergency Medicine, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83301, Taiwan 5 Department of Neurology, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83301, Taiwan 6 Center of Parkinson’s Disease, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83301, Taiwan * Correspondence: [email protected] (C.-T.K.); [email protected] (T.-K.L.) Abstract: Parkinson’s disease (PD) is the second most common neurodegenerative disease and is characterized by dopaminergic neuronal loss. The exact pathogenesis of PD is complex and not yet completely understood, but research has established the critical role mitochondrial dysfunction plays in the development of PD. As the main producer of cytosolic reactive oxygen species (ROS), mito- chondria are particularly susceptible to oxidative stress once an imbalance between ROS generation and the organelle’s antioxidative system occurs. An overabundance of ROS in the mitochondria can Citation: Kung, H.-C.; Lin, K.-J.; lead to mitochondrial dysfunction and further vicious cycles.