Molecular Mechanisms and Outcomes of Arsenic-Induced Histone Acetylation and Microrna Regulation in Cellular Transformation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functional Compensation Among HMGN Variants Modulates the Dnase I Hypersensitive Sites at Enhancers
Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press Research Functional compensation among HMGN variants modulates the DNase I hypersensitive sites at enhancers Tao Deng,1,12 Z. Iris Zhu,2,12 Shaofei Zhang,1 Yuri Postnikov,1 Di Huang,2 Marion Horsch,3 Takashi Furusawa,1 Johannes Beckers,3,4,5 Jan Rozman,3,5 Martin Klingenspor,6,7 Oana Amarie,3,8 Jochen Graw,3,8 Birgit Rathkolb,3,5,9 Eckhard Wolf,9 Thure Adler,3 Dirk H. Busch,10 Valérie Gailus-Durner,3 Helmut Fuchs,3 Martin Hrabeˇ de Angelis,3,4,5 Arjan van der Velde,2,13 Lino Tessarollo,11 Ivan Ovcherenko,2 David Landsman,2 and Michael Bustin1 1Protein Section, Laboratory of Metabolism, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA; 2Computational Biology Branch, National Center for Biotechnology Information, National Library of Medicine, Bethesda, Maryland 20892, USA; 3German Mouse Clinic, Institute of Experimental Genetics, Helmholtz Zentrum München, German Research Center for Environmental Health, 85764 Neuherberg, Germany; 4Experimental Genetics, Center of Life and Food Sciences Weihenstephan, Technische Universität München, 85354 Freising-Weihenstephan, Germany; 5German Center for Diabetes Research (DZD), 85764 Neuherberg, Germany; 6Molecular Nutritional Medicine, Technische Universität München, 85350 Freising, Germany; 7Center for Nutrition and Food Sciences, Technische Universität München, 85350 Freising, Germany; 8Institute of Developmental Genetics (IDG), 85764 -

Expression of HAT1 and HDAC1, 2, 3 in Diffuse Large B-Cell Lymphomas, Peripheral T-Cell Lymphomas, and NK/T-Cell Lymphomas
The Korean Journal of Pathology 2012; 46: 142-150 ▒ ORIGINAL ARTICLE ▒ http://dx.doi.org/10.4132/KoreanJPathol.2012.46.2.142 Expression of HAT1 and HDAC1, 2, 3 in Diffuse Large B-Cell Lymphomas, Peripheral T-Cell Lymphomas, and NK/T-Cell Lymphomas Soo Kee Min · Young Ho Koh1 Background: It has generally been proven that histone acetylation and deacetylation are involved Yunwoong Park1 · Hyo Jung Kim2 in the malignant transformation. To date, however, this has rarely been studied in cases of malig- Jinwon Seo · Hye-Rim Park nant lymphoma. Methods: We studied nine cases of reactive lymphoid hyperplasia, 78 cases of diffuse large B-cell lymphoma (DLBCL), 13 cases of peripheral T-cell lymphoma, not otherwise 3 4 Seong Jin Cho · In Sun Kim specified (PTCL-NOS), and 13 cases of extranodal NK/T-cell lymphoma, nasal type (NKTCL). Thus, we attempted to elucidate the associations of the degree of the expression of histone acetyltrans- Department of Pathology, Hallym University ferase 1 (HAT1), histone deacetylase (HDAC) 1, HDAC2, and HDAC3 with the clinical behaviors of Sacred Heart Hospital, Hallym University College above malignant lymphomas using the immunohistochemistry and a western blot analysis. of Medicine; 1Ilsong Institute of Life Science, Re- Hallym University; 2Department of Hemato- sults: The degree of the expression of HAT1 was higher in cases of DLBCL, PTCL-NOS or NKT- Oncology, Hallym University Sacred Heart CL as compared with reactive lymphoid hyperplasia (p< 0.05). The degree of the expression of Hospital, Hallym University College of Medicine, HAT1 was correlated with that of HDAC1 in cases of DLBCL or NKTCL (p< 0.05). -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Cancer TNT Ashwin Ram 12/5/2017 Background: Chromatin Writers, Readers, Erasers
Cancer TNT Ashwin Ram 12/5/2017 Background: Chromatin Writers, Readers, Erasers writer effector eg. HAT, HMT reader eg. bromodomain eraser eg. HDAC, KDM The writer HAT1: A known H4 lysine 5,12 di-acetyltransferase writer siHAT1 siControl HAT1 H4 K12Ac H4 K5Ac actin Western blot for histone H4 modifications after control and HAT1 siRNA transfections. HAT1: EGF-stimulated immunoprecipitation specific specific - - HAT1 IgG HAT1 IgG - - R α non R α non EGF: + + + - - - WB: HAT1 Immunoprecipitation / WB to measure HAT1 levels +/- Heatmap of gene expression changes of all human histone EGF acetyltransferases +/- EGF and siRNA treatments shows HAT1 expression is EGF-dependent Working model of HAT1 : The oldest “new” histone acetyltransferase EGF EGFR plasma membrane HAT1 H4 H3 Rbap46/48 a nuclear membrane H4 H2A H3 H2B HAT1 S phase Surprise: HAT1 also binds (a few sites) on chromatin HAT1 ChipSeq signal sits on Hist1 locus on Chromosome 6 Read Depth HAT1 bound sites (zoom) HAT1 ChIP-seq peaks cluster at Read Depth histone H4 promoters. Hist1H2BE Hist1H4D Hist1H3D Hist1H4E Is HAT1 a transcription factor for its substrate (H4)? EGF EGFR plasma membrane HAT1 H4 H3 Rbap46/48 a nuclear membrane H4 H2A H3 H2B HAT1 S phase HAT1 is required for S-phase burst of histone H4 mRNA HAT1 Hist1H4B mRNA level 50 Rbap46 45 shCont-3 H4 40 shHAT1-A7 35 shHAT1-B6 30 25 B6 A7 - - 20 15 shHAT1 shHAT1 shControl 10 Gene actin)Expression (versus 5 HAT1 0 0 2 4 6 8 10 actin hours after release from double thymidine block G1 S G2/M HAT1 loss: Life with less histones EGF -

Recombinant Human HAT1 Protein Catalog Number: ATGP0507
Recombinant human HAT1 protein Catalog Number: ATGP0507 PRODUCT INPORMATION Expression system E.coli Domain 20-341aa UniProt No. O14929 NCBI Accession No. NP_003633 Alternative Names Histone acetyltransferase 1, KAT1, Histone acetyltransferase 1 HAT 1, Histidine aminotransferase 1, Histone acetyltransferase type B catalytic subunit PRODUCT SPECIFICATION Molecular Weight 40.1 kDa (343aa) confirmed by MALDI-TOF Concentration 0.5mg/ml (determined by Bradford assay) Formulation Liquid in. 20mM Tris-HCl buffer (pH 8.0) containing 10% glycerol, 1mM DTT Purity > 90% by SDS-PAGE Tag His-Tag Application SDS-PAGE Storage Condition Can be stored at +2C to +8C for 1 week. For long term storage, aliquot and store at -20C to -80C. Avoid repeated freezing and thawing cycles. BACKGROUND Description HAT1, also known as histone acetyltransferase 1, is a type B histone acetyltransferase (HAT) that is involved in the rapid acetylation of newly synthesized cytoplasmic histones, which are in turn imported into the nucleus for de novo deposition onto nascent DNA chains. Histone acetylation, particularly of histone H4, plays an important role in replication-dependent chromatin assembly. Specifically, this HAT can acetylate soluble but not nucleosomal histone H4 at lysines 5 and 12, and to a lesser degree, histone H2A at lysine 5. Recombinant HAT1 1 Recombinant human HAT1 protein Catalog Number: ATGP0507 protein was expressed in E. coli and purified by using conventional chromatography techniques. Amino acid Sequence MGSSHHHHHH SSGLVPRGSH MKKLAEYKCN TNTAIELKLV RFPEDLENDI RTFFPEYTHQ LFGDDETAFG YKGLKILLYY IAGSLSTMFR VEYASKVDEN FDCVEADDVE GKIRQIIPPG FCTNTNDFLS LLEKEVDFKP FGTLLHTYSV LSPTGGENFT FQIYKADMTC RGFREYHERL QTFLMWFIET ASFIDVDDER WHYFLVFEKY NKDGATLFAT VGYMTVYNYY VYPDKTRPRV SQMLILTPFQ GQGHGAQLLE TVHRYYTEFP TVLDITAEDP SKSYVKLRDF VLVKLCQDLP CFSREKLMQG FNEDMAIEAQ QKFKINKQHA RRVYEILRLL VTD General References Verreault A., et al. -

HAT1 Sirna (Rat)
For research purposes only, not for human use Product Data Sheet HAT1 siRNA (Rat) Catalog # Source Reactivity Applications CRR4463 Synthetic R RNAi Description siRNA to inhibit HAT1 expression using RNA interference Specificity HAT1 siRNA (Rat) is a target-specific 19-23 nt siRNA oligo duplexes designed to knock down gene expression. Form Lyophilized powder Gene Symbol HAT1 Alternative Names Histone acetyltransferase type B catalytic subunit; Histone acetyltransferase 1 Entrez Gene 296501 (Rat) SwissProt Q5M939 (Rat) Purity > 97% Quality Control Oligonucleotide synthesis is monitored base by base through trityl analysis to ensure appropriate coupling efficiency. The oligo is subsequently purified by affinity-solid phase extraction. The annealed RNA duplex is further analyzed by mass spectrometry to verify the exact composition of the duplex. Each lot is compared to the previous lot by mass spectrometry to ensure maximum lot-to-lot consistency. Components We offers pre-designed sets of 3 different target-specific siRNA oligo duplexes of rat HAT1 gene. Each vial contains 5 nmol of lyophilized siRNA. The duplexes can be transfected individually or pooled together to achieve knockdown of the target gene, which is most commonly assessed by qPCR or western blot. Our siRNA oligos are also chemically modified (2’-OMe) at no extra charge for increased stability and enhanced knockdown in vitro and in vivo. Directions for Use We recommends transfection with 100 nM siRNA 48 to 72 hours prior to cell lysis. Application key: E- ELISA, WB- Western blot, -

HMGB1 in Health and Disease R
Donald and Barbara Zucker School of Medicine Journal Articles Academic Works 2014 HMGB1 in health and disease R. Kang R. C. Chen Q. H. Zhang W. Hou S. Wu See next page for additional authors Follow this and additional works at: https://academicworks.medicine.hofstra.edu/articles Part of the Emergency Medicine Commons Recommended Citation Kang R, Chen R, Zhang Q, Hou W, Wu S, Fan X, Yan Z, Sun X, Wang H, Tang D, . HMGB1 in health and disease. 2014 Jan 01; 40():Article 533 [ p.]. Available from: https://academicworks.medicine.hofstra.edu/articles/533. Free full text article. This Article is brought to you for free and open access by Donald and Barbara Zucker School of Medicine Academic Works. It has been accepted for inclusion in Journal Articles by an authorized administrator of Donald and Barbara Zucker School of Medicine Academic Works. Authors R. Kang, R. C. Chen, Q. H. Zhang, W. Hou, S. Wu, X. G. Fan, Z. W. Yan, X. F. Sun, H. C. Wang, D. L. Tang, and +8 additional authors This article is available at Donald and Barbara Zucker School of Medicine Academic Works: https://academicworks.medicine.hofstra.edu/articles/533 NIH Public Access Author Manuscript Mol Aspects Med. Author manuscript; available in PMC 2015 December 01. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Mol Aspects Med. 2014 December ; 0: 1–116. doi:10.1016/j.mam.2014.05.001. HMGB1 in Health and Disease Rui Kang1,*, Ruochan Chen1, Qiuhong Zhang1, Wen Hou1, Sha Wu1, Lizhi Cao2, Jin Huang3, Yan Yu2, Xue-gong Fan4, Zhengwen Yan1,5, Xiaofang Sun6, Haichao Wang7, Qingde Wang1, Allan Tsung1, Timothy R. -

Table S6. Names and Functions of 44 Target Genes and 4 Housekeeping Genes Assessed for Gene Expression Measurements
Table S6. Names and functions of 44 target genes and 4 housekeeping genes assessed for gene expression measurements. Gene Gene name Function Category Reference CD45 CD45 (leukocyte common antigen) Regulation of T-cell and B-cell antigen receptor signaling Adaptive NCBI, UniProt HIVEP2 Human immunodeficiency virus typeI enhancer2 Transcription factor, V(D)J recombination, MHC enhancer binding Adaptive (Diepeveen et al. 2013) HIVEP3 Human immunodeficiency virus typeI enhancer3 Transcription factor, V(D)J recombination, MHC enhancer binding Adaptive (Diepeveen et al. 2013) IgM-lc Immunoglobulin light chain Recognition of antigen or pathogen Adaptive NCBI, UniProt Integ-Bt Integrin-beta 1 Cell signaling and adhesion of immunoglobulin Adaptive NCBI, UniProt Lymph75 Lymphocyte antigen 75 Directs captured antigens to lymphocytes Adaptive (Birrer et al. 2012) Lympcyt Lymphocyte cytosolic protein 2 Positive role in promoting T-cell development and activation Adaptive NCBI, UniProt TAP Tap-binding protein (Tapasin) Transport of antigenic peptides, peptide loading on MHC I Adaptive NCBI, UniProt AIF Allograft inflammation factor Inflammatory responses, allograft rejection, activation of macrophages Innate (Roth et al. 2012) Calrcul Calreticulin Chaperone, promotes phagocytosis and clearance of apoptotic cells Innate NCBI, UniProt Cf Coagulation factor II Blood clotting and inflammation response Innate (Birrer et al. 2012) IL8 Interleukin 8 Neutrophil chemotactic factor, phagocytosis, inflammatory activity Innate NCBI, UniProt Intf Interferon induced transmembrane protein 3 Negative regulation of viral entry into host cell, antiviral response Innate NCBI, UniProt Kin Kinesin Intracellular transport Innate (Roth et al. 2012) LectptI Lectin protein type I Pathogen recognition receptors (C-type lectin type I) Innate NCBI, UniProt LectpII Lectin protein type II Pathogen recognition receptors (C-type lectin type II) Innate NCBI, UniProt Nramp Natural resistance-assoc macrophage protein Macrophage activation Innate (Roth et al. -

Cytogenetics in Oncology and Haematology
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Scope The Atlas of Genetics and Cytogenetics in Oncology and Haematology is a peer reviewed on-line journal in open access, devoted to genes, cytogenetics, and clinical entities in cancer, and cancer-prone diseases. It presents structured review articles ("cards") on genes, leukaemias, solid tumours, cancer-prone diseases, more traditional review articles on these and also on surrounding topics ("deep insights"), case reports in hematology, and educational items in the various related topics for students in Medicine and in Sciences. Editorial correspondance Jean-Loup Huret Genetics, Department of Medical Information, University Hospital F-86021 Poitiers, France tel +33 5 49 44 45 46 or +33 5 49 45 47 67 [email protected] or [email protected] Staff Mohammad Ahmad, Mélanie Arsaban, Mikael Cordon, Isabelle Dabin, Marie-Christine Jacquemot-Perbal, Maureen Labarussias, Anne Malo, Catherine Morel-Pair, Laurent Rassinoux, Sylvie Yau Chun Wan - Senon, Alain Zasadzinski. Philippe Dessen is the Database Director, and Alain Bernheim the Chairman of the on-line version (Gustave Roussy Institute – Villejuif – France). The Atlas of Genetics and Cytogenetics in Oncology and Haematology (ISSN 1768-3262) is published 12 times a year by ARMGHM, a non profit organisation, and by the INstitute for Scientific and Technical Information of the French National Center for Scientific Research (INIST-CNRS) since 2008. The Atlas is hosted by INIST-CNRS (http://www.inist.fr) http://AtlasGeneticsOncology.org The PDF version of the Atlas of Genetics and Cytogenetics in Oncology and Haematology is a reissue of the original articles published in collaboration with the Institute for Scientific and Technical Information (INstitut de l’Information Scientifique et Technique - INIST) of the French National Center for Scientific Research (CNRS) on its electronic publishing platform I-Revues. -
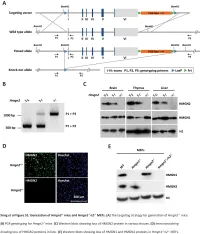
Supplemental Data.Pdf
Table S1. Summary of sequencing results A. DNase‐seq data % Align % Mismatch % >=Q30 bases Sample ID # Reads % unique reads (PF) Rate (PF) (PF) WT_1 84,301,522 86.76 0.52 93.34 96.30 WT_2 98,744,222 84.97 0.51 93.75 89.94 Hmgn1‐/‐_1 79,620,656 83.94 0.86 90.58 88.65 Hmgn1‐/‐_2 62,673,782 84.13 0.87 91.30 89.18 Hmgn2‐/‐_1 87,734,440 83.49 0.71 91.81 90.00 Hmgn2‐/‐_2 82,498,808 83.25 0.69 92.73 90.66 Hmgn1‐/‐n2‐/‐_1 71,739,638 68.51 2.31 81.11 89.22 Hmgn1‐/‐n2‐/‐_2 74,113,682 68.19 2.37 81.16 86.57 B. ChIP‐seq data Histone % Align % Mismatch % >=Q30 % unique Genotypes # Reads marks (PF) Rate (PF) bases (PF) reads H3K4me1 100670054 92.99 0.28 91.21 87.29 H3K4me3 67064272 91.97 0.35 89.11 27.15 WT H3K27ac 90,340,242 93.57 0.28 95.02 89.80 input 111,292,572 78.24 0.55 96.07 86.99 H3K4me1 84598176 92.34 0.33 91.2 81.69 H3K4me3 90032064 92.19 0.44 88.76 15.81 Hmgn1‐/‐n2‐/‐ H3K27ac 86,260,526 93.40 0.29 94.94 87.49 input 78,142,334 78.47 0.56 95.82 81.11 C. MNase‐seq data % Mismatch % >=Q30 bases % unique Sample ID # Reads % Align (PF) Rate (PF) (PF) reads WT_1_Extensive 45,232,694 55.23 1.49 90.22 81.73 WT_1_Limited 105,460,950 58.03 1.39 90.81 79.62 WT_2_Extensive 40,785,338 67.34 1.06 89.76 89.60 WT_2_Limited 105,738,078 68.34 1.05 90.29 85.96 Hmgn1‐/‐n2‐/‐_1_Extensive 117,927,050 55.74 1.49 89.50 78.01 Hmgn1‐/‐n2‐/‐_1_Limited 61,846,742 63.76 1.22 90.57 84.55 Hmgn1‐/‐n2‐/‐_2_Extensive 137,673,830 60.04 1.30 89.28 78.99 Hmgn1‐/‐n2‐/‐_2_Limited 45,696,614 62.70 1.21 90.71 85.52 D. -

A Yeast Phenomic Model for the Influence of Warburg Metabolism on Genetic Buffering of Doxorubicin Sean M
Santos and Hartman Cancer & Metabolism (2019) 7:9 https://doi.org/10.1186/s40170-019-0201-3 RESEARCH Open Access A yeast phenomic model for the influence of Warburg metabolism on genetic buffering of doxorubicin Sean M. Santos and John L. Hartman IV* Abstract Background: The influence of the Warburg phenomenon on chemotherapy response is unknown. Saccharomyces cerevisiae mimics the Warburg effect, repressing respiration in the presence of adequate glucose. Yeast phenomic experiments were conducted to assess potential influences of Warburg metabolism on gene-drug interaction underlying the cellular response to doxorubicin. Homologous genes from yeast phenomic and cancer pharmacogenomics data were analyzed to infer evolutionary conservation of gene-drug interaction and predict therapeutic relevance. Methods: Cell proliferation phenotypes (CPPs) of the yeast gene knockout/knockdown library were measured by quantitative high-throughput cell array phenotyping (Q-HTCP), treating with escalating doxorubicin concentrations under conditions of respiratory or glycolytic metabolism. Doxorubicin-gene interaction was quantified by departure of CPPs observed for the doxorubicin-treated mutant strain from that expected based on an interaction model. Recursive expectation-maximization clustering (REMc) and Gene Ontology (GO)-based analyses of interactions identified functional biological modules that differentially buffer or promote doxorubicin cytotoxicity with respect to Warburg metabolism. Yeast phenomic and cancer pharmacogenomics data were integrated to predict differential gene expression causally influencing doxorubicin anti-tumor efficacy. Results: Yeast compromised for genes functioning in chromatin organization, and several other cellular processes are more resistant to doxorubicin under glycolytic conditions. Thus, the Warburg transition appears to alleviate requirements for cellular functions that buffer doxorubicin cytotoxicity in a respiratory context. -

Characterizing and Selectively Targeting RNF20 Defects Within Colorectal Cancer Cells by Brent John Guppy a Thesis Submitted To
Characterizing and Selectively Targeting RNF20 Defects Within Colorectal Cancer Cells by Brent John Guppy A Thesis submitted to the Faculty of Graduate Studies of The University of Manitoba In partial fulfillment of the requirements of the degree of Doctor of Philosophy Department of Biochemistry and Medical Genetics University of Manitoba Winnipeg Copyright © 2016 by Brent John Guppy ABSTRACT By 2030, the global colorectal cancer burden is projected to approximately double. This highlights the immediate need to expand our understanding of the etiological origins of colorectal cancer, so that novel therapeutic strategies can be identified and validated. The putative tumor suppressor gene RNF20 encodes a histone H2B mono-ubiquitin ligase and has been found altered/mutated in colorectal and numerous other cancer types. Several studies suggest that RNF20, and by extension mono-ubiquitinated histone H2B (H2Bub1), play important roles in maintaining genome stability in human cells. Indeed, hypomorphic RNF20 expression and/or function have been shown to underlie several phenotypes consistent with genome instability, making aberrant RNF20 biology a potential driver in oncogenesis. Through an evolutionarily conserved trans-histone pathway, RNF20 and H2Bub1 have been shown to modulate downstream di-methylation events at lysines 4 (H3K4me2) and 79 (H3K79me2) of histone H3. Accordingly, understanding the biology associated with RNF20, H2Bub1, H3K4me2, and H3K79me2 is an essential preliminary step towards understanding the etiological origins of cancer-associated RNF20 alterations and identifying a novel therapeutic strategy to selectively kill RNF20-deficient cancers. In this thesis, I employ single-cell imaging, and multiple biochemical techniques to investigate the spatial and temporal patterning and characterize the biology of RNF20, H2Bub1, H3K4me2 and H3K79me2 throughout the cell cycle.