Package Insert
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Shelf-Stable Food Safety
United States Department of Agriculture Food Safety and Inspection Service Food Safety Information PhotoDisc Shelf-Stable Food Safety ver since man was a hunter-gatherer, he has sought ways to preserve food safely. People living in cold climates Elearned to freeze food for future use, and after electricity was invented, freezers and refrigerators kept food safe. But except for drying, packing in sugar syrup, or salting, keeping perishable food safe without refrigeration is a truly modern invention. What does “shelf stable” Foods that can be safely stored at room temperature, or “on the shelf,” mean? are called “shelf stable.” These non-perishable products include jerky, country hams, canned and bottled foods, rice, pasta, flour, sugar, spices, oils, and foods processed in aseptic or retort packages and other products that do not require refrigeration until after opening. Not all canned goods are shelf stable. Some canned food, such as some canned ham and seafood, are not safe at room temperature. These will be labeled “Keep Refrigerated.” How are foods made In order to be shelf stable, perishable food must be treated by heat and/ shelf stable? or dried to destroy foodborne microorganisms that can cause illness or spoil food. Food can be packaged in sterile, airtight containers. All foods eventually spoil if not preserved. CANNED FOODS What is the history of Napoleon is considered “the father” of canning. He offered 12,000 French canning? francs to anyone who could find a way to prevent military food supplies from spoiling. Napoleon himself presented the prize in 1795 to chef Nicholas Appert, who invented the process of packing meat and poultry in glass bottles, corking them, and submerging them in boiling water. -
FROM DNA to BEER Date Lesson Plan: Acquired and Passive Immunity Class Period
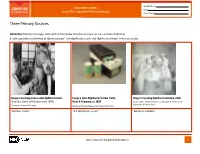
Student Name FROM DNA TO BEER Date Lesson Plan: Acquired and Passive Immunity Class Period Three Primary Sources Instruction: Examine the images and read their titles below. Based on what you can learn and infer from them: 1. Write your inferred definition of “diphtheria toxin,” “anti-diphtheritic serum,” and “diphtheria antitoxin” in the table below. Image 1. Injecting a horse with diphtheria toxin, Image 2. Anti-Diphtheritic Serum, Parke, Image 3. Injecting diphtheria antitoxin, 1895 New York City Health Department, 1940s Davis & Company, ca. 1898 Courtesy The Historical Medical Library of The College of Physicians of Philadelphia Courtesy Library of Congress Courtesy National Museum of American History “diphtheria toxin”: “anti-diphtheritic serum”: “diphtheria antitoxin”: http://www.nlm.nih.gov/fromdnatobeer 1 Student Name FROM DNA TO BEER Date Lesson Plan: Acquired and Passive Immunity Class Period Three Primary Sources 2. Describe or draw how the three images may be related. http://www.nlm.nih.gov/fromdnatobeer 2 FROM DNA TO BEER Lesson Plan: Acquired and Passive Immunity Teacher’s Three Primary Sources Instruction: Examine the images and read their titles below. Based on what you can learn and infer from them: 1. Write your inferred definition of “diphtheria toxin,” “anti-diphtheritic serum,” and “diphtheria antitoxin” in the table below. Image 1. Injecting a horse with diphtheria toxin, Image 2. Anti-Diphtheritic Serum, Parke, Image 3. Injecting diphtheria antitoxin, 1895 New York City Health Department, 1940s Davis & Company, -

Botulism Fact Sheet
Botulism Fact Sheet 1. What is botulism? - Botulism is a rare but serious paralytic illness caused by a nerve toxin (poison) produced by the bacterium Clostridium botulinum. There are three main kinds of botulism. (a) Foodborne botulism is caused by eating foods that contain the botulism toxin. (b) Wound botulism is caused by toxin produced from a wound infected with Clostridium botulinum. (c) Infant botulism is caused when Clostridium botulinum bacteria grow in the immature infant intestines and release toxin. All forms of botulism can be fatal and are considered medical emergencies. Foodborne botulism can be especially dangerous because widespread illness can occur from eating a contaminated food. 2. What is Clostridium botulinum? - Clostridium botulinum belongs to is a group of bacteria commonly found in soil. They grow best in low oxygen conditions. The bacteria form spores which allow them to survive in a dormant state in the soil until exposed to conditions that can support their growth. In low oxygen conditions, the growing bacteria release botulinum toxin. There are seven types of botulism toxin designated by the letters A through G; only types A, B, E and F cause illness in humans. 3. How common is botulism? - In the United States an average of 45 cases are reported each year. Of these, approximately 15% are foodborne, 65% are infant botulism, and the rest are wound botulism. Outbreaks of foodborne botulism involving two or more persons occur most years and usually are caused by eating improperly processed home-canned foods. The number of Foodborne and Infant botulism cases has changed little in recent years, but wound botulism has increased because of the use of black-tar heroin. -

Botulism Guide for Health Care Professionals
Botulism Guide for Health Care Professionals This information requires knowledgeable interpretation and is intended primarily for use by health care workers and facilities/organizations providing health care including pharmacies, hospitals, long-term care homes, community-based health care service providers and pre-hospital emergency services. Population and Public Health Division Ministry of Health and Long-Term Care March 2017 AT A GLANCE A Quick Response Guide to Botulism Botulism – The treatment of botulism is guided by clinical diagnosis The initial diagnosis of botulism should be based on a history of recent exposure, consistent clinical symptoms and elimination of other illnesses in the differential. Treatment should not wait for laboratory confirmation. All treatment and management decisions should be made based on clinical diagnosis. Initial Presentation and evaluation of signs and symptoms There are several clinically distinct forms of botulism. All forms produce the same neurological signs and symptoms of symmetrical cranial nerve palsies followed by descending, symmetric flaccid paralysis of voluntary muscles, which may progress to respiratory compromise and death. Additional symptoms (e.g., gastrointestinal signs in foodborne cases) may also be seen in some forms. Read more on the disease on page 2 Reading the section on Differential Diagnosis and the referenced articles will assist with making the diagnosis of botulism – you will find this on page 3 Place a request for Botulinum Antitoxin (BAT) or BabyBIG® Ministry of Health and Long-Term Care (ministry) staff will arrange for the shipment of BAT. Information on ordering BAT and BabyBIG (BabyBIG has a different ordering process) is on page 5 Laboratory Diagnosis and Specimen Collection Clinical specimens must be obtained prior to administering treatment with botulinum antitoxin. -

Botulism Manual
Preface This report, which updates handbooks issued in 1969, 1973, and 1979, reviews the epidemiology of botulism in the United States since 1899, the problems of clinical and laboratory diagnosis, and the current concepts of treatment. It was written in response to a need for a comprehensive and current working manual for epidemiologists, clinicians, and laboratory workers. We acknowledge the contributions in the preparation of this review of past and present physicians, veterinarians, and staff of the Foodborne and Diarrheal Diseases Branch, Division of Bacterial and Mycotic Diseases (DBMD), National Center for Infectious Diseases (NCID). The excellent review of Drs. K.F. Meyer and B. Eddie, "Fifty Years of Botulism in the United States,"1 is the source of all statistical information for 1899-1949. Data for 1950-1996 are derived from outbreaks reported to CDC. Suggested citation Centers for Disease Control and Prevention: Botulism in the United States, 1899-1996. Handbook for Epidemiologists, Clinicians, and Laboratory Workers, Atlanta, GA. Centers for Disease Control and Prevention, 1998. 1 Meyer KF, Eddie B. Fifty years of botulism in the U.S. and Canada. George Williams Hooper Foundation, University of California, San Francisco, 1950. 1 Dedication This handbook is dedicated to Dr. Charles Hatheway (1932-1998), who served as Chief of the National Botulism Surveillance and Reference Laboratory at CDC from 1975 to 1997. Dr. Hatheway devoted his professional life to the study of botulism; his depth of knowledge and scientific integrity were known worldwide. He was a true humanitarian and served as mentor and friend to countless epidemiologists, research scientists, students, and laboratory workers. -

Anasakid Nematodes and Fish
Anasakid Nematodes and Fish Why should we be concerned? Anasakid nematodes are small, round worms. The larvae (immature worms) and adult worms can sometimes can be seen in the organs or flesh of fish, but sometimes not. What can we do to keep safe from these nematodes? It is not known how often there are nematodes in fish in Nunavut. Gut fish as soon as they are killed. If you do not If the larvae in the fish are not killed, gut the fish right after they can cause sickness in humans who harvesting, freeze the fish eat the fish. right away. Cook the fish. Cooking will kill the worms so they can not cause sickness. How does it make If you want to eat the people sick? fish uncooked, freeze it first. Freezing fish will People who eat fish kill both immature and with nematode larvae adult worms. Freeze fish can have tingling in the throat. People for 7 days at -20°C before can also develop stomach ulcers (which eating raw. cause pain in the stomach). -20C˚ 7 days -20˚ It is not known if drying fish kills the nematodes Botulism Why should we be concerned? The botulism bacteria Botulism bacteria can Botulism sickness are found naturally in make toxins if they do not can be very serious in walrus and seal. The have air. The toxins from humans and can even bacteria may also be the bacteria can cause a cause death. found in whale. sickness in humans called “botulism”. How does it make What can we do to keep safe people sick? from botulism? Bacteria can grow and Remove the stomach and make toxins if they do intestines of seal, whale and 4˚ 4˚C or colder not have air. -

(ACIP) General Best Guidance for Immunization
Appendix 1: Glossary Adverse event. An undesirable medical condition that occurs following vaccination which might be truly caused by a vaccine, or it might be pure coincidence. Adverse reaction. An undesirable medical condition that has been demonstrated to be caused by a vaccine. Evidence for the causal relation is usually obtained through randomized clinical trials, controlled epidemiologic studies, isolation of the vaccine strain from the pathogenic site, or recurrence of the condition with repeated vaccination (i.e., rechallenge); synonyms include side effect and adverse effect. Adjuvant. A vaccine component distinct from the antigen that enhances the immune response to the antigen. Antitoxin. A solution of antibodies against a toxin. Antitoxin can be derived from either human (e.g., tetanus immune globulin) or animal (usually equine) sources (e.g., diphtheria and botulism antitoxin). Antitoxins are used to confer passive immunity and for treatment. Hyperimmune globulin (specific). Special preparations obtained from blood plasma from donor pools preselected for a high antibody content against a specific antigen (e.g., hepatitis B immune globulin, varicella-zoster immune globulin, rabies immune globulin, tetanus immune globulin, vaccinia immune globulin, cytomegalovirus immune globulin, botulism immune globulin). Immune globulin. A sterile solution containing antibodies, which are usually obtained from human blood. It is obtained by cold ethanol fractionation of large pools of blood plasma and contains 15%-18% protein. Intended for intramuscular administration, immune globulin is primarily indicated for routine maintenance of immunity among certain immunodeficient persons and for passive protection against measles and hepatitis A. General Best Practice Guidelines for Immunization: Appendix 1: Glossary 189 Immunobiologic. Antigenic substances (e.g., vaccines and toxoids) or antibody- containing preparations (e.g., globulins and antitoxins) from human or animal donors. -

Bacterial Foodborne and Diarrheal Disease National Case Surveillance
Bacterial Foodborne and Diarrheal Disease National Case Surveillance Annual Report, 2003 Enteric Diseases Epidemiology Branch Division of Foodborne, Bacterial and Mycotic Diseases National Center for Zoonotic, Vectorborne and Enteric Diseases Centers for Disease Control and Prevention The Bacterial Foodborne and Diarrheal Disease National Case Surveillance is published by the Enteric Diseases Epidemiology Branch, Division of Foodborne, Bacterial and Mycotic Diseases, National Center for Zoonotic, Vectorborne and Enteric Diseases, Centers for Disease Control and Prevention, Atlanta, GA 30333 SUGGESTED CITATION Centers for Disease Control and Prevention. Bacterial Foodborne and Diarrheal Disease National Case Surveillance. Annual Report, 2003. Atlanta Centers for Disease Control and Prevention; 2005: pg. Nos - 2 - Contents Executive Summary……………………………………………………………………………… - 4- Expanded Surveillance Summaries of Selected Pathogens and Diseases, 2003………………… -10- Botulism…………………………………………………………………………………. -10- Non-O157 Shiga toxin-producing Escherichia coli………………………………………-18- Salmonella………………………………………………………………………………...-22- Shigella……………………………………………………………………………………-28- Vibrio……………………………………………………………………………………...-33- Surveillance Data Sources and Background……………………………………………………... -40- National Notifiable Diseases Surveillance System and the National Electronic Telecommunications System for Surveillance…………………………………………… -40- Public Health Laboratory Information System…………………………………………... -41- Limitations common to NETSS and PHLIS…………………………………………….. -

Clostridium Botulinum1 Keith R
FSHN0406 Preventing Foodborne Illness: Clostridium botulinum1 Keith R. Schneider, Renée M. Goodrich Schneider, Ploy Kurdmongkoltham, and Bruna Bertoldi2 This fact sheet is part of a series that discusses foodborne potent neurotoxin that causes botulism, a serious paralytic pathogens of interest to food handlers, processors, retailers, condition that can lead to death. and consumers. There are seven types of C. botulinum (A, B, C, D, E, F, and What is Clostridium botulinum? G), each distinguished by the production of serologically distinct toxins. Of the seven types, A, B, E, and rarely F Clostridium botulinum is the bacterium that causes can cause botulism in humans, while types C and D cause botulism. Clostridium botulinum is a Gram-positive, slightly botulism in animals and birds. Type G was identified in curved, motile, anaerobic, rod-shaped bacterium that 1970 but has not been determined as a cause of botulism in produces heat-resistant endospores. These endospores, humans or animals (FDA 2012; Sobel 2005). which are very resistant to a number of environmental stresses, such as heat and high acid, can become activated in anaerobic environments, low acidity (pH > 4.6), high How is Clostridium botulinum moisture content, and in temperatures ranging from 40°F transmitted? to 250°F (4°C to 121°C) (Sobel et al. 2004). In hostile The CDC categorizes human botulism cases into five environmental conditions, the heat-resistant spores enable transmission categories: foodborne, infant, wound, adult the bacteria to survive for extended periods of time in a intestinal toxemia, and iatrogenic botulism. (CDC 2017a). dormant state until conditions become more favorable. -

Diphtheria. In: Epidemiology and Prevention of Vaccine
Diphtheria Anna M. Acosta, MD; Pedro L. Moro, MD, MPH; Susan Hariri, PhD; and Tejpratap S.P. Tiwari, MD Diphtheria is an acute, bacterial disease caused by toxin- producing strains of Corynebacterium diphtheriae. The name Diphtheria of the disease is derived from the Greek diphthera, meaning ● Described by Hippocrates in ‘leather hide.’ The disease was described in the 5th century 5th century BCE BCE by Hippocrates, and epidemics were described in the ● Epidemics described in 6th century AD by Aetius. The bacterium was first observed 6th century in diphtheritic membranes by Edwin Klebs in 1883 and cultivated by Friedrich Löffler in 1884. Beginning in the early ● Bacterium first observed in 1900s, prophylaxis was attempted with combinations of toxin 1883 and cultivated in 1884 and antitoxin. Diphtheria toxoid was developed in the early ● Diphtheria toxoid developed 7 1920s but was not widely used until the early 1930s. It was in 1920s incorporated with tetanus toxoid and pertussis vaccine and became routinely used in the 1940s. Corynebacterium diphtheria Corynebacterium diphtheriae ● Aerobic gram-positive bacillus C. diphtheriae is an aerobic, gram-positive bacillus. ● Toxin production occurs Toxin production (toxigenicity) occurs only when the when bacillus is infected bacillus is itself infected (lysogenized) by specific viruses by corynebacteriophages (corynebacteriophages) carrying the genetic information for carrying tox gene the toxin (tox gene). Diphtheria toxin causes the local and systemic manifestations of diphtheria. ● Four biotypes: gravis, intermedius, mitis, and belfanti C. diphtheriae has four biotypes: gravis, intermedius, mitis, ● All isolates should be tested and belfanti. All biotypes can become toxigenic and cause for toxigenicity severe disease. -

Current Medical Literature American J. Digestive Diseases, Fort Wayne, Ind. Am. J. Roentgenol. & Rad. Therapy, Springfield
American Journal of Public Health, New York Current Medical Literature 33:925-1042 (Aug.) 1943 National Board of Health 1879-1883. W. G. Smillie.—p. 925. Preventive Medicine Program of United States Army. J. S. Simmons. AMERICAN —p. 931. Home Methods and Their Effect on Quality The Association to of Drying Palatability, Cooking library lends periodicals members the Association and Nutritive Value of Foods. Esther L. 941. and to Batchelder.—p. individual subscribers in continental United States and Canada Blood and Malaria Parasite Staining with Eosin Azure Méthylène Blue for a of be borrowed a time. period three days. Three journals may at Methods. R. D. 948. Periodicals are available from 1933 to date. for issues Lillie.—p. Requests of Radio Habits of Attend earlier date cannot be filled. should be Listening Mothers Who Well Baby Clinics. Requests accompanied by L. Murray and C. E. 952. cover if one and 18 if three Margaret Turner.—p. stamps to postage (6 cents cents periodicals Surveys of Nutrition of Populations: 2. Protein Nutrition of Rural are requested). Periodicals the American Medical Asso¬ published by Population in Middle Tennessee. J. B. E. W. ciation are not available for but can be on Youmans, Patton, lending supplied purchase W. R. Ruth Kern and Ruth 955. order. as a rule are the of can be Sutton, Steinkamp.—p. Reprints property authors and Field for Health Education Personnel. Minnie obtained for permanent possession only from them. Experience Krueger Oed. —p. 965. Titles marked with an asterisk (*) are abstracted below. Dehydration Procedures and Their Effect on Vitamin Retention. -

Humoral Immune Response Evaluation in Horses Vaccinated with Recombinant Clostridium Perfringens Toxoids Alpha and Beta for 12 Months
toxins Article Humoral Immune Response Evaluation in Horses Vaccinated with Recombinant Clostridium perfringens Toxoids Alpha and Beta for 12 Months Nayra F. Q. R. Freitas 1, Denis Y. Otaka 1, Cleideanny C. Galvão 1, Dayane M. de Almeida 1, Marcos R. A. Ferreira 2, Clóvis Moreira Júnior 2 , Marina M. M. H. Hidalgo 3, Fabricio R. Conceição 2 and Felipe M. Salvarani 1,* 1 Instituto de Medicina Veterinária, Universidade Federal do Pará, Castanhal CEP 68740-970, Brazil; [email protected] (N.F.Q.R.F.); [email protected] (D.Y.O.); [email protected] (C.C.G.); [email protected] (D.M.d.A.) 2 Centro de Desenvolvimento Tecnológico, Núcleo de Biotecnologia, Universidade Federal de Pelotas, Rio Grande do Sul CEP 96160-000, Brazil; [email protected] (M.R.A.F.); [email protected] (C.M.J.); [email protected] (F.R.C.) 3 Faculdade de Veterinária, Universidade Federal de Pelotas, Rio Grande do Sul CEP 96160-000, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-91-99-31-73-503 Abstract: In horses, Clostridium perfringens is associated with acute and fatal enterocolitis, which is caused by a beta toxin (CPB), and myonecrosis, which is caused by an alpha toxin (CPA). Although the most effective way to prevent these diseases is through vaccination, specific clostridial vaccines for horses against C. perfringens are not widely available. The aim of this study was to pioneer the immunization of horses with three different concentrations (100, 200 and 400 µg) of C. perfringens Citation: Freitas, N.F.Q.R.; Otaka, recombinant alpha (rCPA) and beta (rCPB) proteins, as well as to evaluate the humoral immune D.Y.; Galvão, C.C.; de Almeida, D.M.; response over 360 days.