Identification of Genes Associated with Tumorigenesis and Metastatic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

Lysophosphatidic Acid and Its Receptors: Pharmacology and Therapeutic Potential in Atherosclerosis and Vascular Disease
JPT-107404; No of Pages 13 Pharmacology & Therapeutics xxx (2019) xxx Contents lists available at ScienceDirect Pharmacology & Therapeutics journal homepage: www.elsevier.com/locate/pharmthera Lysophosphatidic acid and its receptors: pharmacology and therapeutic potential in atherosclerosis and vascular disease Ying Zhou a, Peter J. Little a,b, Hang T. Ta a,c, Suowen Xu d, Danielle Kamato a,b,⁎ a School of Pharmacy, University of Queensland, Pharmacy Australia Centre of Excellence, Woolloongabba, QLD 4102, Australia b Department of Pharmacy, Xinhua College of Sun Yat-sen University, Tianhe District, Guangzhou 510520, China c Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, St Lucia, QLD 4072, Australia d Aab Cardiovascular Research Institute, Department of Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY 14642, USA article info abstract Available online xxxx Lysophosphatidic acid (LPA) is a collective name for a set of bioactive lipid species. Via six widely distributed G protein-coupled receptors (GPCRs), LPA elicits a plethora of biological responses, contributing to inflammation, Keywords: thrombosis and atherosclerosis. There have recently been considerable advances in GPCR signaling especially Lysophosphatidic acid recognition of the extended role for GPCR transactivation of tyrosine and serine/threonine kinase growth factor G-protein coupled receptors receptors. This review covers LPA signaling pathways in the light of new information. The use of transgenic and Atherosclerosis gene knockout animals, gene manipulated cells, pharmacological LPA receptor agonists and antagonists have Gproteins fi β-arrestins provided many insights into the biological signi cance of LPA and individual LPA receptors in the progression Transactivation of atherosclerosis and vascular diseases. -

The Autotaxin–Lysophosphatidic Acid Axis Modulates Histone Acetylation and Gene Expression During Oligodendrocyte Differentiation
The Journal of Neuroscience, August 12, 2015 • 35(32):11399–11414 • 11399 Cellular/Molecular The Autotaxin–Lysophosphatidic Acid Axis Modulates Histone Acetylation and Gene Expression during Oligodendrocyte Differentiation Natalie A. Wheeler,1 James A. Lister,2 and Babette Fuss1 Departments of 1Anatomy and Neurobiology and 2Human and Molecular Genetics, Virginia Commonwealth University School of Medicine, Richmond, Virginia 23298 During development, oligodendrocytes (OLGs), the myelinating cells of the CNS, undergo a stepwise progression during which OLG progenitors, specified from neural stem/progenitor cells, differentiate into fully mature myelinating OLGs. This progression along the OLG lineage is characterized by well synchronized changes in morphology and gene expression patterns. The latter have been found to be particularly critical during the early stages of the lineage, and they have been well described to be regulated by epigenetic mechanisms, especially by the activity of the histone deacetylases HDAC1 and HDAC2. The data presented here identify the extracellular factor autotaxin (ATX) as a novel upstream signal modulating HDAC1/2 activity and gene expression in cells of the OLG lineage. Using the zebrafish as an in vivo model system as well as rodent primary OLG cultures, this functional property of ATX was found to be mediated by its lysophospholipase D (lysoPLD) activity, which has been well characterized to generate the lipid signaling molecule lysophosphatidic acid (LPA). More specifically, the lysoPLD activity of ATX was found to modulate HDAC1/2 regulated gene expression during a time window coinciding with the transition from OLG progenitor to early differentiating OLG. In contrast, HDAC1/2 regulated gene expression during the transition from neural stem/progenitor to OLG progenitor appeared unaffected by ATX and its lysoPLD activity. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Antibody Response Cell Antigen Receptor Signaling And
Lysophosphatidic Acid Receptor 5 Inhibits B Cell Antigen Receptor Signaling and Antibody Response This information is current as Jiancheng Hu, Shannon K. Oda, Kristin Shotts, Erin E. of September 24, 2021. Donovan, Pamela Strauch, Lindsey M. Pujanauski, Francisco Victorino, Amin Al-Shami, Yuko Fujiwara, Gabor Tigyi, Tamas Oravecz, Roberta Pelanda and Raul M. Torres J Immunol 2014; 193:85-95; Prepublished online 2 June 2014; Downloaded from doi: 10.4049/jimmunol.1300429 http://www.jimmunol.org/content/193/1/85 Supplementary http://www.jimmunol.org/content/suppl/2014/05/31/jimmunol.130042 http://www.jimmunol.org/ Material 9.DCSupplemental References This article cites 63 articles, 17 of which you can access for free at: http://www.jimmunol.org/content/193/1/85.full#ref-list-1 Why The JI? Submit online. by guest on September 24, 2021 • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2014 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Lysophosphatidic Acid Receptor 5 Inhibits B Cell Antigen Receptor Signaling and Antibody Response Jiancheng Hu,*,1,2 Shannon K. -

Microgenomics of Ameloblastoma
RESEARCH REPORTS Biological P. DeVilliers1, C. Suggs2, D. Simmons2, V. Murrah3, and J.T. Wright2* Microgenomics of 1Department of Pathology, University of Alabama at Ameloblastoma Birmingham; 2Department of Pediatric Dentistry, CB #7450, and 3Department of Oral Pathology, University of North Carolina at Chapel Hill, NC 27599, USA; *corresponding author, [email protected] J Dent Res 90(4):463-469, 2011 ABSTRACT INTRODUCTION Gene expression profiles of human ameloblastoma microdissected cells were characterized with the meloblastoma is an aggressive tumor of odontogenic epithelial origin, purpose of identifying genes and their protein Awith devastating morbidity if left untreated, due to its unlimited growth products that could be targeted as diagnostic and potential. It is characterized by a high rate of recurrence (up to 70%, depend- prognostic markers as well as for potential thera- ing on the treatment modality) and potential to undergo malignant transfor- peutic interventions. Five formalin-fixed, decalci- mation and to metastasize (up to 2% of cases). Malignant ameloblastoma is fied, paraffin-embedded samples of ameloblastoma defined as a histologically benign-appearing ameloblastoma with metastasis were subjected to laser capture microdissection, (Goldenberg et al., 2004; Cardoso et al., 2009). Surgical resection is the treat- linear mRNA amplification, and hybridization to ment of choice, which can cause further morbidity to the craniofacial com- oligonucleotide human 41,000 RNA arrays and plex, with loss of function and esthetics (Olasoji and Enwere, 2003). compared with universal human reference RNA, Gene expression profiling of tumor cell populations has advanced our to determine the gene expression signature. understanding of the pathogenesis of human tumors (Naderi et al., 2004). -

Molecular Pathways Associated with the Nutritional Programming of Plant-Based Diet Acceptance in Rainbow Trout Following an Early Feeding Exposure Mukundh N
Molecular pathways associated with the nutritional programming of plant-based diet acceptance in rainbow trout following an early feeding exposure Mukundh N. Balasubramanian, Stéphane Panserat, Mathilde Dupont-Nivet, Edwige Quillet, Jérôme Montfort, Aurélie Le Cam, Françoise Médale, Sadasivam J. Kaushik, Inge Geurden To cite this version: Mukundh N. Balasubramanian, Stéphane Panserat, Mathilde Dupont-Nivet, Edwige Quillet, Jérôme Montfort, et al.. Molecular pathways associated with the nutritional programming of plant-based diet acceptance in rainbow trout following an early feeding exposure. BMC Genomics, BioMed Central, 2016, 17 (1), pp.1-20. 10.1186/s12864-016-2804-1. hal-01346912 HAL Id: hal-01346912 https://hal.archives-ouvertes.fr/hal-01346912 Submitted on 19 Jul 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Balasubramanian et al. BMC Genomics (2016) 17:449 DOI 10.1186/s12864-016-2804-1 RESEARCH ARTICLE Open Access Molecular pathways associated with the nutritional programming of plant-based diet acceptance in rainbow trout following an early feeding exposure Mukundh N. Balasubramanian1, Stephane Panserat1, Mathilde Dupont-Nivet2, Edwige Quillet2, Jerome Montfort3, Aurelie Le Cam3, Francoise Medale1, Sadasivam J. Kaushik1 and Inge Geurden1* Abstract Background: The achievement of sustainable feeding practices in aquaculture by reducing the reliance on wild-captured fish, via replacement of fish-based feed with plant-based feed, is impeded by the poor growth response seen in fish fed high levels of plant ingredients. -
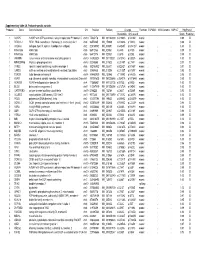
Supplementary Table 2A. Proband-Specific Variants Proband
Supplementary table 2A. Proband-specific variants Proband Gene Gene full name Chr Position RefSeqChange Function ESP6500 1000 Genome SNP137 PolyPhen2 Nucleotide Amino acid Score Prediction 1 AGAP5 ArfGAP with GTPase domain, ankyrin repeat and PH domain 5 chr10 75435178 NM_001144000 c.G1240A p.V414M exonic . 0.99 D 1 REXO1L1 REX1, RNA exonuclease 1 homolog (S. cerevisiae)-like 1 chr8 86574465 NM_172239 c.A1262G p.Y421C exonic . 0.98 D 1 COL4A3 collagen, type IV, alpha 3 (Goodpasture antigen) chr2 228169782 NM_000091 c.G4235T p.G1412V exonic . 1.00 D 1 KIAA1586 KIAA1586 chr6 56912133 NM_020931 c.C44G p.A15G exonic . 0.99 D 1 KIAA1586 KIAA1586 chr6 56912176 NM_020931 c.C87G p.D29E exonic . 0.99 D 1 JAKMIP3 Janus kinase and microtubule interacting protein 3 chr10 133955524 NM_001105521 c.G1574C p.G525A exonic . 1.00 D 1 MPHOSPH8 M-phase phosphoprotein 8 chr13 20240685 NM_017520 c.C2140T p.L714F exonic . 0.97 D 1 SYNE1 spectrin repeat containing, nuclear envelope 1 chr6 152783902 NM_033071 c.A2242T p.N748Y exonic . 0.97 D 1 CAND2 cullin-associated and neddylation-dissociated 2 (putative) chr3 12858835 NM_012298 c.C2125T p.H709Y exonic . 0.95 D 1 TDRD9 tudor domain containing 9 chr14 104460909 NM_153046 c.T1289G p.V430G exonic . 0.96 D 2 ASPM asp (abnormal spindle) homolog, microcephaly associated (Drosophila)chr1 197091625 NM_001206846 c.G3491A p.R1164H exonic . 0.99 D 2 ADAM29 ADAM metallopeptidase domain 29 chr4 175896951 NM_001130705 c.A275G p.D92G exonic . 1.00 D 2 BCO2 beta-carotene oxygenase 2 chr11 112087019 NM_001256398 c.G1373A p.G458E exonic . -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Identification of a Gene Coding for a Protein Possessing Shared Tumor Epitopes Capable of Inducing HLA-A24-Restricted Cytotoxic T Lymphocytes in Cancer Patients1
[CANCER RESEARCH 59, 4056–4063, August 15, 1999] Identification of a Gene Coding for a Protein Possessing Shared Tumor Epitopes Capable of Inducing HLA-A24-restricted Cytotoxic T Lymphocytes in Cancer Patients1 Damu Yang, Masanobu Nakao, Shigeki Shichijo, Teruo Sasatomi, Hideo Takasu, Hajime Matsumoto, Kazunori Mori, Akihiro Hayashi, Hideaki Yamana, Kazuo Shirouzu, and Kyogo Itoh2 Cancer Vaccine Development Division, Kurume University Research Center for Innovative Cancer Therapy [D. Y., M. N., K. I.], and Departments of Surgery [A. H., H. Y., K. S.], Immunology [S. S., T. S., H. T., H. M., K. I.], and Otolaryngology [K. M.], Kurume University School of Medicine, Kurume, 830-0011, Japan ABSTRACT we report a gene encoding epitopes that are capable of inducing CTLs in PBMCs of patients with SCCs and adenocarcinomas. Genes encoding tumor epitopes that are capable of inducing CTLs against adenocarcinomas and squamous cell carcinomas, two major hu- man cancers histologically observed in various organs, have rarely been MATERIALS AND METHODS identified. Here, we report a new gene from cDNA of esophageal cancer cells that encodes a shared tumor antigen recognized by HLA-A2402- Generation of HLA-A2402-restricted CTLs. HLA-A2402-restricted and restricted and tumor-specific CTLs. The sequence of this gene is almost tumor-specific CTLs were established from the PBMCs of an esophageal identical to that of the KIAA0156 gene, which has been registered in cancer patient (HLA-A2402/A2601) by the standard method of mixed lym- phocyte tumor cell culture, -

Structural and Biochemical Changes Underlying a Keratoderma-Like Phenotype in Mice Lacking Suprabasal AP1 Transcription Factor Function
Citation: Cell Death and Disease (2015) 6, e1647; doi:10.1038/cddis.2015.21 OPEN & 2015 Macmillan Publishers Limited All rights reserved 2041-4889/15 www.nature.com/cddis Structural and biochemical changes underlying a keratoderma-like phenotype in mice lacking suprabasal AP1 transcription factor function EA Rorke*,1, G Adhikary2, CA Young2, RH Rice3, PM Elias4, D Crumrine4, J Meyer4, M Blumenberg5 and RL Eckert2,6,7,8 Epidermal keratinocyte differentiation on the body surface is a carefully choreographed process that leads to assembly of a barrier that is essential for life. Perturbation of keratinocyte differentiation leads to disease. Activator protein 1 (AP1) transcription factors are key controllers of this process. We have shown that inhibiting AP1 transcription factor activity in the suprabasal murine epidermis, by expression of dominant-negative c-jun (TAM67), produces a phenotype type that resembles human keratoderma. However, little is understood regarding the structural and molecular changes that drive this phenotype. In the present study we show that TAM67-positive epidermis displays altered cornified envelope, filaggrin-type keratohyalin granule, keratin filament, desmosome formation and lamellar body secretion leading to reduced barrier integrity. To understand the molecular changes underlying this process, we performed proteomic and RNA array analysis. Proteomic study of the corneocyte cross-linked proteome reveals a reduction in incorporation of cutaneous keratins, filaggrin, filaggrin2, late cornified envelope precursor proteins, hair keratins and hair keratin-associated proteins. This is coupled with increased incorporation of desmosome linker, small proline-rich, S100, transglutaminase and inflammation-associated proteins. Incorporation of most cutaneous keratins (Krt1, Krt5 and Krt10) is reduced, but incorporation of hyperproliferation-associated epidermal keratins (Krt6a, Krt6b and Krt16) is increased. -

CDH12 Cadherin 12, Type 2 N-Cadherin 2 RPL5 Ribosomal
5 6 6 5 . 4 2 1 1 1 2 4 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 A A A A A A A A A A A A A A A A A A A A C C C C C C C C C C C C C C C C C C C C R R R R R R R R R R R R R R R R R R R R B , B B B B B B B B B B B B B B B B B B B , 9 , , , , 4 , , 3 0 , , , , , , , , 6 2 , , 5 , 0 8 6 4 , 7 5 7 0 2 8 9 1 3 3 3 1 1 7 5 0 4 1 4 0 7 1 0 2 0 6 7 8 0 2 5 7 8 0 3 8 5 4 9 0 1 0 8 8 3 5 6 7 4 7 9 5 2 1 1 8 2 2 1 7 9 6 2 1 7 1 1 0 4 5 3 5 8 9 1 0 0 4 2 5 0 8 1 4 1 6 9 0 0 6 3 6 9 1 0 9 0 3 8 1 3 5 6 3 6 0 4 2 6 1 0 1 2 1 9 9 7 9 5 7 1 5 8 9 8 8 2 1 9 9 1 1 1 9 6 9 8 9 7 8 4 5 8 8 6 4 8 1 1 2 8 6 2 7 9 8 3 5 4 3 2 1 7 9 5 3 1 3 2 1 2 9 5 1 1 1 1 1 1 5 9 5 3 2 6 3 4 1 3 1 1 4 1 4 1 7 1 3 4 3 2 7 6 4 2 7 2 1 2 1 5 1 6 3 5 6 1 3 6 4 7 1 6 5 1 1 4 1 6 1 7 6 4 7 e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m