Complete Genome Sequence of the Endophytic Bacterium Cellulosimicrobium Sp. JZ28 Isolated from Root Endosphere of the Perennial
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bioprospecting of Elite Plant Growth-Promoting Bacteria for the Maize Crop
Acta Scientiarum http://periodicos.uem.br/ojs/acta ISSN on-line: 1807-8621 Doi: 10.4025/actasciagron.v42i1.44364 AGRICULTURE MICROBIOLOGY Bioprospecting of elite plant growth-promoting bacteria for the maize crop Angela Cristina Ikeda1* , Daiani Cristina Savi1, Mariangela Hungria2, Vanessa Kava1, Chirlei Glienke1 and Lygia Vitória Galli-Terasawa1 1Universidade Federal do Paraná, Rua XV de Novembro, 1299, 80060-000, Curitiba, Paraná, Brazil. 2Empresa Brasileira de Pesquisa Agropecuária, Embrapa Soja, Londrina, Paraná, Brazil. *Author for Correspondence. E-mail: [email protected] ABSTRACT. The use of plant growth-promoting bacteria (PGPB), which aims to replace chemical fertilizers and biological control, is a goal for achieving agriculture sustainability. In this scenario, our goal was to identify and evaluate the potential of bacteria isolated from maize roots to promote plant growth and be used as inoculants. We evaluated 173 bacterial strains isolated from the maize (Zea mays L.) rhizosphere for the properties of their PGPB in vitro. Twelve strains were positive for siderophores, indole acetic acid (IAA) production, biological nitrogen fixation (BNF), and phosphate solubilization. Sequence analysis of 16S rRNA identified these strains as belonging to the genera Cellulosimicrobium, Stenotrophomonas, Enterobacter, and Bacillus. The elite strains were evaluated under greenhouse conditions upon the inoculation of two maize hybrids, ATL100 and KWX628. The ability of the isolates to promote plant growth was dependent on the maize genotype; Enterobacter sp. LGMB208 showed the best ability to promote growth of hybrid ATL100, while Enterobacter sp. strains LGMB125, LGMB225, and LGMB274 and Cellulosimicrobium sp. strain LGMB239 showed the best ability to promote growth of hybrid KWX628. The results highlight the potential of bacterial genera little explored as maize PGPB but indicate the need to investigate their interactions with different plant genotypes. -

A Case Report of the Differential Diagnosis of Cellulosimicrobium Cellulans-Infected Endocarditis Combined with Intracranial
Zhang et al. BMC Infectious Diseases (2020) 20:893 https://doi.org/10.1186/s12879-020-05559-6 CASE REPORT Open Access A case report of the differential diagnosis of Cellulosimicrobium cellulans-infected endocarditis combined with intracranial infection by conventional blood culture and second-generation sequencing Huifang Zhang1†, Chunyan He2†, Rui Tian1 and Ruilan Wang1* Abstract Background: Cellulosimicrobium cellulans is a gram-positive filamentous bacterium found primarily in soil and sewage that rarely causes human infection, especially in previously healthy adults, but when it does, it often indicates a poor prognosis. Case presentation: We report a case of endocarditis and intracranial infection caused by C. cellulans in a 52-year- old woman with normal immune function and no implants in vivo. The patient started with a febrile headache that progressed to impaired consciousness after 20 days, and she finally died after treatment with vancomycin combined with rifampicin. C. cellulans was isolated from her blood cultures for 3 consecutive days after her admission; however, there was only evidence of C. cellulans sequences for two samples in the second-generation sequencing data generated from her peripheral blood, which were ignored by the technicians. No C. cellulans bands were detected in her cerebrospinal fluid by second-generation sequencing. Conclusions: Second-generation sequencing seems to have limitations for certain specific strains of bacteria. Keywords: Cellulosimicrobium cellulans, Second-generation sequencing, Infectious endocarditis, Case report Background report a case of endocarditis and intracranial infection Cellulosimicrobium cellulans, formerly known as Oersko- caused by C. cellulans in a 52-year-old woman with via xanthineolytica, is a gram-positive filamentous bac- normal immune function and no implants in vivo. -

Infections Due to Cellulosimicrobium Species: Case Report and Literature
Rivero et al. BMC Infectious Diseases (2019) 19:816 https://doi.org/10.1186/s12879-019-4440-2 CASEREPORT Open Access Infections due to Cellulosimicrobium species: case report and literature review María Rivero1,2*, Javier Alonso3, María Fernanda Ramón3, Nancy Gonzales3, Ana Pozo3, Itxaso Marín3, Ana Navascués4 and Regina Juanbeltz2,5,6 Abstract Background: Cellulosimicrobium species, formely known as Oerskovia species, are gram-positive bacilli belonging to the order Actinomycetales. They rarely cause human infections. The genus comprises two pathogenic species in humans: C. cellulans and C. funkei. Based on a case report, we provide a review of the literature of infections caused by Cellulosimicrobium/Oerskovia, in order to improve our knowledge of this unusual infection. Case presentation: An 82-year-old woman with aortic prosthetic valve presented to the hospital with fever and heart failure. Further work up revealed the diagnosis of C. cellulans infective endocarditis (IE). The strain was identified by MALDI-TOF MS, API Coryne and 16S rRNA sequencing. The patient was deemed not to be an operative candidate and died despite the antibiotic therapy 35 days after admission. Conclusions: Reviewing cases of Cellulosimicrobium species infections and communicating the successful and unsuccessful clinical experiences can assist future healthcare providers. Our case and those previously reported indicate that Cellulosimicrobium species usually infect immunocompromised patients or foreign body carriers. The most frequent pattern of infection is central venous catheter related bacteremia. The optimal treatment should include foreign body removal and valve surgery should be considered in case of IE. Keywords: Cellulosimicrobium, Oerskovia, Central venous catheter, Endocarditis, Foreign body Background caused by these organisms, in order to improve our Cellulosimicrobium species are gram-positive bacilli knowledge of this unusual infection. -
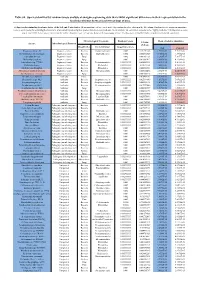
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp. -

Of the 16 Strains Obtained from Salty Soil in Mongolia, Six Strains Were
African Journal of Microbiology Research Vol. 7(4), pp. 298-308, 22 January, 2013 Available online at http://www.academicjournals.org/AJMR DOI: 10.5897/AJMR12.498 ISSN 1996 0808 ©2013 Academic Journals Full Length Research Paper Isolation, classification, phylogenetic analysis and scanning electron microscopy of halophilic, halotolerant and alkaliphilic actinomycetes isolated from hypersaline soil Ismet Ara1,2*, D. Daram4, T. Baljinova4, H. Yamamura5, W. N. Hozzein3, M. A. Bakir1, M. Suto2 and K. Ando2 1Department of Botany and Microbiology, College of Science, King Saud University, P. O. Box 22452, Riyadh-11495, Kingdom of Saudi Arabia. 2Biotechnology Development Center, Department of Biotechnology (DOB), National Institute of Technology and Evaluation (NITE), Kazusakamatari 2-5-8, Kisarazu, Chiba, 292-0818, Japan. 3Bioproducts Research Chair (BRC), College of Science, King Saud University. 4Division of Applied Biological Sciences, Interdisciplinary Graduate School of Medicine and Engineering, University of Yamanashi, Takeda-4, Kofu 400-8511, Japan. Accepted 9 January, 2013 Actinomycetes were isolated by plating of serially diluted samples onto humic acid-vitamin agar prepared with or without NaCl and characterized by physiological and phylogenetical studies. A total of 16 strains were isolated from hypersaline soil in Mongolia. All strains showed alkaliphilic characteristics and were able to abundantly grow in media with pH 9.0. Six strains were halotolerant actinomycetes (0 to 12.0% NaCl) and 10 strains were moderately halophiles (3.0 to 12.0% NaCl). Most strains required moderately high salt (5.0 to 10.0%) for optimal growth but were able to grow at lower NaCl concentrations. Among the 16 strains, 5 were able to grow at 45°C. -

1 Possible Drivers in Endophyte Diversity and Transmission in The
Possible Drivers in Endophyte Diversity and Transmission in the Tomato Plant Bacterial Microbiome Thesis Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Ana María Vázquez, B.S. Graduate Program in Plant Pathology The Ohio State University 2020 Thesis Committee Dr. María Soledad Benítez-Ponce, Advisor Dr. Christine Sprunger Dr. Jonathan M. Jacobs 1 Copyrighted by Ana María Vázquez 2020 2 Abstract It has been documented that beneficial plant-associated bacteria have contributed to disease suppression, growth promotion, and tolerance to abiotic stresses. Advances in high-throughput sequencing have allowed an increase in research regarding bacterial endophytes, which are microbes that colonize the interior of plants without causing disease. Practices associated with minimizing the use of off-farm resources, such as reduced tillage regimes and crop rotations, can cause shifts in plant-associated bacteria and its surrounding agroecosystem. Integrated crop–livestock systems are an option that can provide environmental benefits by implementing diverse cropping systems, incorporating perennial and legume forages and adding animal manure through grazing livestock. It has been found that crop-livestock systems can increase soil quality and fertility, reduce cost of herbicide use and improve sustainability, especially for farmers in poorer areas of the world. This work explores how crop-livestock systems that integrate chicken rotations can impact tomato plant growth, as well as soil and endophytic bacterial communities. Tomato plants were subjected to greenhouse and field studies where biomass was assessed, and bacterial communities were characterized through culture- dependent and -independent approaches. -

Reclassification of Cellulomonas Cellulans (Stackebrandt and Keddie 1986) As Cellulosimicrobium Cellulans Gen. Nov., Comb. Nov
International Journal of Systematic and Evolutionary Microbiology (2001), 51, 1007–1010 Printed in Great Britain Reclassification of Cellulomonas cellulans NOTE (Stackebrandt and Keddie 1986) as Cellulosimicrobium cellulans gen. nov., comb. nov. DSMZ-Deutsche Sammlung Peter Schumann, Norbert Weiss and Erko Stackebrandt von Mikroorganismen und Zellkulturen GmbH, Mascheroder Weg 1b, 38124 Braunschweig, Author for correspondence: Erko Stackebrandt. Tel: j49 531 2616352. Fax: j49 531 2616418. Germany e-mail: erko!dsmz.de Phylogenetic analysis of 16S rDNA provides evidence that Cellulomonas cellulans branches outside the phylogenetic confines of the genus Cellulomonas. The distinct phylogenetic and chemotaxonomic status of Cellulomonas cellulans as a phylogenetic neighbour of the genus Promicromonospora, justifies the description of a new genus for which the name Cellulosimicrobium gen. nov. with the type species Cellulosimicrobium cellulans comb. nov. is proposed. Keywords: Cellulomonas, Cellulosimicrobium gen. nov., Cellulosimicrobium cellulans comb. nov. The genus Cellulomonas is a member of the suborder nature nucleotides, were described as families Micrococcineae, order Actinomycetales, class Actino- (Stackebrandt et al., 1997). Among these are the bacteria. Most members show cellulolytic activity and families Cellulomonadaceae, embracing the genus one species, Cellulomonas hominis (Funke et al., 1996) Cellulomonas, and Promicromonosporaceae, contain- has been isolated from clinical material. Besides those ing the genus Promicromonospora. Since then, several species that have been members of the genus since new genera of this suborder have been described its original description (Bergey et al., 1923), i.e. (Groth et al., 1997a, b, 1999; Martin et al., 1997) Cellulomonas flavigena, Cellulomonas biazotea, which has changed the phylogenetic position of a few Cellulomonas cellasea, Cellulomonas gelida, Cellulo- taxa and stabilized other taxa. -

Assessment of Bioremediation Potential of Cellulosimicrobium Sp
Microbiol. Biotechnol. Lett. (2019), 47(2), 269–277 http://dx.doi.org/10.4014/mbl.1808.08006 pISSN 1598-642X eISSN 2234-7305 Microbiology and Biotechnology Letters Assessment of Bioremediation Potential of Cellulosimicrobium sp. for Treatment of Multiple Heavy Metals Tushar Bhati, Rahul Gupta, Nisha Yadav, Ruhi Singh, Antra Fuloria, Aafrin Waziri, Sayan Chatterjee, and Ram Singh Purty* University School of Biotechnology, Guru Gobind Singh Indraprastha University, Sec-16C, Dwarka, New Delhi, India Received: August 16, 2018 / Revised: November 4, 2018 / Accepted: November 7, 2018 In the present study, we have studied the bioremediating capability of bacterial strain against six heavy metals. The strain was isolated from river Yamuna, New Delhi which is a very rich repository of bioremedi- ating flora and fauna. The strain was found to be Gram positive as indicated by Gram staining. The strain was characterized using 16s rRNA gene sequencing and the BlastN result showed its close resemblance with the Cellulosimicrobium sp. As each treatment has its own toxicity eliciting expression of different fac- tors, we observed varied growth characteristics of the bacterial isolate and its protein content in response to different heavy metals. The assessment of its bioremediation capability showed that the strain Cellulo- simicrobium sp. has potential to consume or sequester the six heavy metals in this study in the following order iron > lead > zinc > cooper > nickel > cadmium. Thus, the strain Cellulosimicrobium sp. isolated in the present study can be a good model system to understand the molecular mechanism behind its bioreme- diating capabilities under multiple stress conditions. Keywords: Cellulosimicrobium sp, bioremediation, multiple heavy metals tolerant, Yamuna river Introduction cell membrane, disrupt cellular function, and destroy the DNA structure. -

Antimicrobial Activity of Springtails' Bacteria
VU Research Portal Diversity and functions of bacteria associated with springtails Agamennone, V. 2018 document version Publisher's PDF, also known as Version of record Link to publication in VU Research Portal citation for published version (APA) Agamennone, V. (2018). Diversity and functions of bacteria associated with springtails. General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. E-mail address: [email protected] Download date: 26. Sep. 2021 Chapter 4 – Antimicrobial activity in culturable gut microbial communities of springtails Agamennone V, Roelofs D, van Straalen NM, Janssens TKS. Journal of Applied Microbiology 2018; doi: 10.1111/jam.13899 65 Chapter 4 4.1. Abstract Aims The rise of antibiotic resistance pushes the pharmaceutical industry to continually search for substances with new structures and novel mechanisms of action. Many environmental niches are still to be explored as sources of antimicrobials. In this paper we assess the antimicrobial potential of gut microbes of springtails, soil invertebrates which live in a microbe-dominated environment and are known to be tolerant to entomopathogenic microorganisms. -

Isolation and Characterization of Heavy Metal Resistant Cellulosimicrobium Sp. from Paper Mill Polluted Soil Introduction Mate
International Journal of Bioassays ISSN: 2278-778X CODEN: IJBNHY Original Research Article OPEN ACCESS ISOLATION AND CHARACTERIZATION OF HEAVY METAL RESISTANT CELLULOSIMICROBIUM SP. FROM PAPER MILL POLLUTED SOIL Dhritiman Chanda1*, Sharma GD2, Jha DK3, Hijri M4 and F Al-Otaibi4 1Microbiology Laboratory, Department of Life Sciences and Bioinformatics, Assam University, Silchar, India 2Bilaspur University, Chattisgarh, India 3Department of Botany, Gauhati University, Gauhati, India 4Institut de Recherche en Biologie Vegetale, University de Montreal, Montreal, Canada. Received for publication: December 11, 2014; Accepted: December 21, 2014 Abstract: A Gram-positive, rod-shaped, yellow-pigmented bacterium was isolated from the polluted soil of paper mill contaminated with various heavy metals. The strain was tested for its resistance to different heavy metals (Ni, Cu, Zn and Cd) by the growth in nutrient broth tubes containing various concentrations of (0.1, 0.5, 2.0, 4.0 mM). The relative growths (%) at 2mM concentration were observed as Ni (23.52%)>Zn (20.58%)>Cu (19.20%) > Cd (15.81 %). The heavy metal resistant in the isolated strain was found to be Ni>Zn>Cu>Cd at higher concentrations. The strain showed positive activity towards urease, nitrate, citrate utilization, methyl red, Malonate utilization, starch amylase and showed negative activity against ONPG, H2S production, phynylalanine, Lysine utilization, Voges Proskauer’s (VP) test, catalase and oxidase activity. The strain was found susceptible to various antibiotics Vancomycin, Streptomycin, Rifamycin Amikacin and Ciprofloxacin. In silico study was conducted to understand the major evolutionary relationship among the different strains of Cellulosimicrobium species of nucleotide sequence of 16s ribosomal RNA with the isolated strain (KC602297) of Cellulosimicrobium sp. -

Culturable Microorganisms Associated with Sea Cucumbers and Microbial Natural Products
marine drugs Review Culturable Microorganisms Associated with Sea Cucumbers and Microbial Natural Products Lei Chen * , Xiao-Yu Wang, Run-Ze Liu and Guang-Yu Wang * Department of Bioengineering, School of Marine Science and Technology, Harbin Institute of Technology at Weihai, Weihai 264209, China; [email protected] (X.-Y.W.); [email protected] (R.-Z.L.) * Correspondence: [email protected] or [email protected] (L.C.); [email protected] or [email protected] (G.-Y.W.); Tel.: +86-631-5687076 (L.C.); +86-631-5682925 (G.-Y.W.) Abstract: Sea cucumbers are a class of marine invertebrates and a source of food and drug. Numerous microorganisms are associated with sea cucumbers. Seventy-eight genera of bacteria belonging to 47 families in four phyla, and 29 genera of fungi belonging to 24 families in the phylum Ascomycota have been cultured from sea cucumbers. Sea-cucumber-associated microorganisms produce diverse secondary metabolites with various biological activities, including cytotoxic, antimicrobial, enzyme- inhibiting, and antiangiogenic activities. In this review, we present the current list of the 145 natural products from microorganisms associated with sea cucumbers, which include primarily polyketides, as well as alkaloids and terpenoids. These results indicate the potential of the microorganisms associated with sea cucumbers as sources of bioactive natural products. Keywords: sea cucumber; bioactivity; diversity; microorganism; polyketides; alkaloids Citation: Chen, L.; Wang, X.-Y.; Liu, 1. Introduction R.-Z.; Wang, G.-Y. Culturable Sea cucumbers are marine invertebrates that belong to the class Holothuroidea of the Microorganisms Associated with Sea phylum Echinodermata. Globally, there are about 1500 species of sea cucumbers [1], which Cucumbers and Microbial Natural are divided into three subclasses: Aspidochirotacea, Apodacea, and Dendrochirotacea, and Products. -

Inhibitory Bacteria Reduce Fungi on Early Life Stages of Endangered Colorado Boreal Toads (Anaxyrus Boreas)
The ISME Journal (2016) 10, 934–944 © 2016 International Society for Microbial Ecology All rights reserved 1751-7362/16 www.nature.com/ismej ORIGINAL ARTICLE Inhibitory bacteria reduce fungi on early life stages of endangered Colorado boreal toads (Anaxyrus boreas) Jordan G Kueneman1, Douglas C Woodhams1,3, Will Van Treuren2,4, Holly M Archer1, Rob Knight2,5 and Valerie J McKenzie1 1Department of Ecology and Evolutionary Biology, University of Colorado, Boulder, CO, USA and 2BioFrontiers Institute, University of Colorado, Boulder, CO, USA Increasingly, host-associated microbiota are recognized to mediate pathogen establishment, providing new ecological perspectives on health and disease. Amphibian skin-associated microbiota interact with the fungal pathogen, Batrachochytrium dendrobatidis (Bd), but little is known about microbial turnover during host development and associations with host immune function. We surveyed skin microbiota of Colorado’s endangered boreal toads (Anaxyrus boreas), sampling 181 toads across four life stages (tadpoles, metamorphs, subadults and adults). Our goals were to (1) understand variation in microbial community structure among individuals and sites, (2) characterize shifts in communities during development and (3) examine the prevalence and abundance of known Bd-inhibitory bacteria. We used high-throughput 16S and 18S rRNA gene sequencing (Illumina MiSeq) to characterize bacteria and microeukaryotes, respectively. Life stage had the largest effect on the toad skin microbial community, and site and Bd presence also contributed. Proteobacteria dominated tadpole microbial communities, but were later replaced by Actinobacteria. Microeukar- yotes on tadpoles were dominated by the classes Alveolata and Stramenopiles, while fungal groups replaced these groups after metamorphosis. Using a novel database of Bd-inhibitory bacteria, we found fewer Bd-inhibitory bacteria in post-metamorphic stages correlated with increased skin fungi, suggesting that bacteria have a strong role in early developmental stages and reduce skin- associated fungi.