Deoxythymidylate Kinase, DTYMK, Is a Novel Gene for Mitochondrial DNA
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Polyphosphate-Dependent Synthesis of ATP and ADP by the Family-2 Polyphosphate Kinases in Bacteria
Polyphosphate-dependent synthesis of ATP and ADP by the family-2 polyphosphate kinases in bacteria Boguslaw Noceka, Samvel Kochinyanb, Michael Proudfootb, Greg Brownb, Elena Evdokimovaa,b, Jerzy Osipiuka, Aled M. Edwardsa,b, Alexei Savchenkoa,b, Andrzej Joachimiaka,c,1, and Alexander F. Yakuninb,1 aMidwest Center for Structural Genomics and Structural Biology Center, Department of Biosciences, Argonne National Laboratory, 9700 South Cass Avenue, Building 202, Argonne, IL 60439; bBanting and Best Department of Medical Research, University of Toronto, Toronto, ON, Canada M5G 1L6; and cDepartment of Biochemistry and Molecular Biology, University of Chicago, 920 East 58th Street, Chicago, IL 60637 Edited by Robert Haselkorn, University of Chicago, Chicago, IL, and approved September 30, 2008 (received for review August 1, 2008) Inorganic polyphosphate (polyP) is a linear polymer of tens or hun- exopolysaccharide essential for the virulence of P. aeruginosa (12). dreds of phosphate residues linked by high-energy bonds. It is found In the social slime mold Dictyostelium discoideum, a different PPK in all organisms and has been proposed to serve as an energy source was found (DdPPK2) with sequence and properties similar to that in a pre-ATP world. This ubiquitous and abundant biopolymer plays of actin-related proteins (Arps) (14). DdPPK2 is a complex of 3 numerous and vital roles in metabolism and regulation in prokaryotes Arps (Arp1, Arp2, and Arpx) and resembles the actin family in and eukaryotes, but the underlying molecular mechanisms for most molecular weight, sequence, and filamentous structure. Remark- activities of polyP remain unknown. In prokaryotes, the synthesis and ably, DdPPK2 can polymerize into an actin-like filament concurrent utilization of polyP are catalyzed by 2 families of polyP kinases, PPK1 with the synthesis of polyP (14). -

Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’S Biological Network Based on Systematic Pharmacology
ORIGINAL RESEARCH published: 25 June 2021 doi: 10.3389/fphar.2021.601846 Exploring the Regulatory Mechanism of Hedysarum Multijugum Maxim.-Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’s Biological Network Based on Systematic Pharmacology Kailin Yang 1†, Liuting Zeng 1†, Anqi Ge 2†, Yi Chen 1†, Shanshan Wang 1†, Xiaofei Zhu 1,3† and Jinwen Ge 1,4* Edited by: 1 Takashi Sato, Key Laboratory of Hunan Province for Integrated Traditional Chinese and Western Medicine on Prevention and Treatment of 2 Tokyo University of Pharmacy and Life Cardio-Cerebral Diseases, Hunan University of Chinese Medicine, Changsha, China, Galactophore Department, The First 3 Sciences, Japan Hospital of Hunan University of Chinese Medicine, Changsha, China, School of Graduate, Central South University, Changsha, China, 4Shaoyang University, Shaoyang, China Reviewed by: Hui Zhao, Capital Medical University, China Background: Clinical research found that Hedysarum Multijugum Maxim.-Chuanxiong Maria Luisa Del Moral, fi University of Jaén, Spain Rhizoma Compound (HCC) has de nite curative effect on cerebral ischemic diseases, *Correspondence: such as ischemic stroke and cerebral ischemia-reperfusion injury (CIR). However, its Jinwen Ge mechanism for treating cerebral ischemia is still not fully explained. [email protected] †These authors share first authorship Methods: The traditional Chinese medicine related database were utilized to obtain the components of HCC. The Pharmmapper were used to predict HCC’s potential targets. Specialty section: The CIR genes were obtained from Genecards and OMIM and the protein-protein This article was submitted to interaction (PPI) data of HCC’s targets and IS genes were obtained from String Ethnopharmacology, a section of the journal database. -

Stem Cells® Original Article
® Stem Cells Original Article Properties of Pluripotent Human Embryonic Stem Cells BG01 and BG02 XIANMIN ZENG,a TAKUMI MIURA,b YONGQUAN LUO,b BHASKAR BHATTACHARYA,c BRIAN CONDIE,d JIA CHEN,a IRENE GINIS,b IAN LYONS,d JOSEF MEJIDO,c RAJ K. PURI,c MAHENDRA S. RAO,b WILLIAM J. FREEDa aCellular Neurobiology Research Branch, National Institute on Drug Abuse, Department of Health and Human Services (DHHS), Baltimore, Maryland, USA; bLaboratory of Neuroscience, National Institute of Aging, DHHS, Baltimore, Maryland, USA; cLaboratory of Molecular Tumor Biology, Division of Cellular and Gene Therapies, Center for Biologics Evaluation and Research, Food and Drug Administration, Bethesda, Maryland, USA; dBresaGen Inc., Athens, Georgia, USA Key Words. Embryonic stem cells · Differentiation · Microarray ABSTRACT Human ES (hES) cell lines have only recently been compared with pooled human RNA. Ninety-two of these generated, and differences between human and mouse genes were also highly expressed in four other hES lines ES cells have been identified. In this manuscript we (TE05, GE01, GE09, and pooled samples derived from describe the properties of two human ES cell lines, GE01, GE09, and GE07). Included in the list are genes BG01 and BG02. By immunocytochemistry and reverse involved in cell signaling and development, metabolism, transcription polymerase chain reaction, undifferenti- transcription regulation, and many hypothetical pro- ated cells expressed markers that are characteristic of teins. Two focused arrays designed to examine tran- ES cells, including SSEA-3, SSEA-4, TRA-1-60, TRA-1- scripts associated with stem cells and with the 81, and OCT-3/4. Both cell lines were readily main- transforming growth factor-β superfamily were tained in an undifferentiated state and could employed to examine differentially expressed genes. -

Chromosomal Microarray Analysis in Turkish Patients with Unexplained Developmental Delay and Intellectual Developmental Disorders
177 Arch Neuropsychitry 2020;57:177−191 RESEARCH ARTICLE https://doi.org/10.29399/npa.24890 Chromosomal Microarray Analysis in Turkish Patients with Unexplained Developmental Delay and Intellectual Developmental Disorders Hakan GÜRKAN1 , Emine İkbal ATLI1 , Engin ATLI1 , Leyla BOZATLI2 , Mengühan ARAZ ALTAY2 , Sinem YALÇINTEPE1 , Yasemin ÖZEN1 , Damla EKER1 , Çisem AKURUT1 , Selma DEMİR1 , Işık GÖRKER2 1Faculty of Medicine, Department of Medical Genetics, Edirne, Trakya University, Edirne, Turkey 2Faculty of Medicine, Department of Child and Adolescent Psychiatry, Trakya University, Edirne, Turkey ABSTRACT Introduction: Aneuploids, copy number variations (CNVs), and single in 39 (39/123=31.7%) patients. Twelve CNV variant of unknown nucleotide variants in specific genes are the main genetic causes of significance (VUS) (9.75%) patients and 7 CNV benign (5.69%) patients developmental delay (DD) and intellectual disability disorder (IDD). were reported. In 6 patients, one or more pathogenic CNVs were These genetic changes can be detected using chromosome analysis, determined. Therefore, the diagnostic efficiency of CMA was found to chromosomal microarray (CMA), and next-generation DNA sequencing be 31.7% (39/123). techniques. Therefore; In this study, we aimed to investigate the Conclusion: Today, genetic analysis is still not part of the routine in the importance of CMA in determining the genomic etiology of unexplained evaluation of IDD patients who present to psychiatry clinics. A genetic DD and IDD in 123 patients. diagnosis from CMA can eliminate genetic question marks and thus Method: For 123 patients, chromosome analysis, DNA fragment analysis alter the clinical management of patients. Approximately one-third and microarray were performed. Conventional G-band karyotype of the positive CMA findings are clinically intervenable. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

Genome-Wide Linkage Analysis of Human Auditory Cortical Activation Suggests Distinct Loci on Chromosomes 2, 3, and 8
The Journal of Neuroscience, October 17, 2012 • 32(42):14511–14518 • 14511 Behavioral/Systems/Cognitive Genome-Wide Linkage Analysis of Human Auditory Cortical Activation Suggests Distinct Loci on Chromosomes 2, 3, and 8 Hanna Renvall,1* Elina Salmela,2,3* Minna Vihla,1 Mia Illman,1 Eira Leinonen,2,3 Juha Kere,2,3,4 and Riitta Salmelin1 1Brain Research Unit and MEG Core, O.V. Lounasmaa Laboratory, Aalto University, FI-00076 Aalto, Finland, 2Department of Medical Genetics, Haartman Institute, and Research Programs Unit, Molecular Medicine, University of Helsinki, FI-00014 Helsinki, Finland, 3Folkha¨lsan Institute of Genetics, FI-00014 Helsinki, Finland, and 4Department of Biosciences and Nutrition, and Science for Life Laboratory, Karolinska Institute, SE-14183 Stockholm, Sweden Neural processes are explored through macroscopic neuroimaging and microscopic molecular measures, but the two levels remain primarily detached. The identification of direct links between the levels would facilitate use of imaging signals as probes of genetic function and, vice versa, access to molecular correlates of imaging measures. Neuroimaging patterns have been mapped for a few isolated genes,chosenbasedontheirconnectionwithaclinicaldisorder.Hereweproposeanapproachthatallowsanunrestricteddiscoveryofthe genetic basis of a neuroimaging phenotype in the normal human brain. The essential components are a subject population that is composed of relatives and selection of a neuroimaging phenotype that is reproducible within an individual and similar between relatives but markedly variable across a population. Our present combined magnetoencephalography and genome-wide linkage study in 212 healthy siblings demonstrates that auditory cortical activation strength is highly heritable and, specifically in the right hemisphere, regulatedoligogenicallywithlinkagestochromosomes2q37,3p12,and8q24.TheidentifiedregionsdelimitascandidategenesTRAPPC9, operating in neuronal differentiation, and ROBO1, regulating projections of thalamocortical axons. -

New Insights on Human Essential Genes Based on Integrated Multi
bioRxiv preprint doi: https://doi.org/10.1101/260224; this version posted February 5, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. New insights on human essential genes based on integrated multi- omics analysis Hebing Chen1,2, Zhuo Zhang1,2, Shuai Jiang 1,2, Ruijiang Li1, Wanying Li1, Hao Li1,* and Xiaochen Bo1,* 1Beijing Institute of Radiation Medicine, Beijing 100850, China. 2 Co-first author *Correspondence: [email protected]; [email protected] Abstract Essential genes are those whose functions govern critical processes that sustain life in the organism. Comprehensive understanding of human essential genes could enable breakthroughs in biology and medicine. Recently, there has been a rapid proliferation of technologies for identifying and investigating the functions of human essential genes. Here, according to gene essentiality, we present a global analysis for comprehensively and systematically elucidating the genetic and regulatory characteristics of human essential genes. We explain why these genes are essential from the genomic, epigenomic, and proteomic perspectives, and we discuss their evolutionary and embryonic developmental properties. Importantly, we find that essential human genes can be used as markers to guide cancer treatment. We have developed an interactive web server, the Human Essential Genes Interactive Analysis Platform (HEGIAP) (http://sysomics.com/HEGIAP/), which integrates abundant analytical tools to give a global, multidimensional interpretation of gene essentiality. bioRxiv preprint doi: https://doi.org/10.1101/260224; this version posted February 5, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Title: a Yeast Phenomic Model for the Influence of Warburg Metabolism on Genetic
bioRxiv preprint doi: https://doi.org/10.1101/517490; this version posted January 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Title Page: 2 3 Title: A yeast phenomic model for the influence of Warburg metabolism on genetic 4 buffering of doxorubicin 5 6 Authors: Sean M. Santos1 and John L. Hartman IV1 7 1. University of Alabama at Birmingham, Department of Genetics, Birmingham, AL 8 Email: [email protected], [email protected] 9 Corresponding author: [email protected] 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 1 bioRxiv preprint doi: https://doi.org/10.1101/517490; this version posted January 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 26 Abstract: 27 Background: 28 Saccharomyces cerevisiae represses respiration in the presence of adequate glucose, 29 mimicking the Warburg effect, termed aerobic glycolysis. We conducted yeast phenomic 30 experiments to characterize differential doxorubicin-gene interaction, in the context of 31 respiration vs. glycolysis. The resulting systems level biology about doxorubicin 32 cytotoxicity, including the influence of the Warburg effect, was integrated with cancer 33 pharmacogenomics data to identify potentially causal correlations between differential 34 gene expression and anti-cancer efficacy. -

Network-Based Analysis of Key Regulatory Genes Implicated in Type
www.nature.com/scientificreports OPEN Network‑based analysis of key regulatory genes implicated in Type 2 Diabetes Mellitus and Recurrent Miscarriages in Turner Syndrome Anam Farooqui1, Alaa Alhazmi2, Shaful Haque3, Naaila Tamkeen4, Mahboubeh Mehmankhah1, Safa Tazyeen1, Sher Ali5 & Romana Ishrat1* The information on the genotype–phenotype relationship in Turner Syndrome (TS) is inadequate because very few specifc candidate genes are linked to its clinical features. We used the microarray data of TS to identify the key regulatory genes implicated with TS through a network approach. The causative factors of two common co‑morbidities, Type 2 Diabetes Mellitus (T2DM) and Recurrent Miscarriages (RM), in the Turner population, are expected to be diferent from that of the general population. Through microarray analysis, we identifed nine signature genes of T2DM and three signature genes of RM in TS. The power‑law distribution analysis showed that the TS network carries scale‑free hierarchical fractal attributes. Through local‑community‑paradigm (LCP) estimation we fnd that a strong LCP is also maintained which means that networks are dynamic and heterogeneous. We identifed nine key regulators which serve as the backbone of the TS network. Furthermore, we recognized eight interologs functional in seven diferent organisms from lower to higher levels. Overall, these results ofer few key regulators and essential genes that we envisage have potential as therapeutic targets for the TS in the future and the animal models studied here may prove useful in the validation of such targets. Te medical systems and scientists throughout the world are under an unprecedented challenge to meet the medical needs of much of the world’s population that are sufering from chromosomal anomalies. -
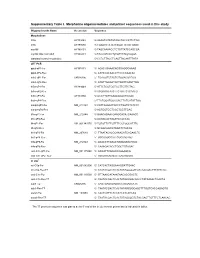
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC -

A Genomic View of Estrogen Actions in Human Breast Cancer Cells by Expression Profiling of the Hormone-Responsive Transcriptome
719 A genomic view of estrogen actions in human breast cancer cells by expression profiling of the hormone-responsive transcriptome Luigi Cicatiello1, Claudio Scafoglio1, Lucia Altucci1, Massimo Cancemi1, Guido Natoli1, Angelo Facchiano2, Giovanni Iazzetti3, Raffaele Calogero4, Nicoletta Biglia6, Michele De Bortoli5,7, Christian Sfiligoi7, Piero Sismondi6,7, Francesco Bresciani1 and Alessandro Weisz1 1Dipartimento di Patologia generale, Seconda Università degli Studi di Napoli, Vico L. De Crecchio 7, 80138 Napoli, Italy 2Istituto di Scienze dell’Alimentazione del Consiglio Nazionale delle Ricerche, Avellino, Italy 3Dipartimento di Genetica, Biologia generale e molecolare, Università di Napoli ‘Federico II’, Napoli, Italy 4Dipartimento di Scienze cliniche e biologiche, Università degli Studi di Torino, Torino, Italy 5Dipartimento di Scienze oncologiche, Università degli Studi di Torino, Torino, Italy 6Dipartimento di Discipline ostetriche e ginecologiche, Università degli Studi di Torino, Torino, Italy 7Laboratorio di Ginecologia oncologica, Istituto per la Ricerca e la Cura del Cancro, Candiolo, Italy (Requests for offprints should be addressed to A Weisz; Email: [email protected]) Abstract Estrogen controls key cellular functions of responsive cells including the ability to survive, replicate, communicate and adapt to the extracellular milieu. Changes in the expression of 8400 genes were monitored here by cDNA microarray analysis during the first 32 h of human breast cancer (BC) ZR-75·1 cell stimulation with a mitogenic dose of 17-estradiol, a timing which corresponds to completion of a full mitotic cycle in hormone-stimulated cells. Hierarchical clustering of 344 genes whose expression either increases or decreases significantly in response to estrogen reveals that the gene expression program activated by the hormone in these cells shows 8 main patterns of gene activation/inhibition. -

The 2Q37-Deletion Syndrome: an Update of the Clinical Spectrum Including Overweight, Brachydactyly and Behavioural Features in 14 New Patients
European Journal of Human Genetics (2013) 21, 602–612 & 2013 Macmillan Publishers Limited All rights reserved 1018-4813/13 www.nature.com/ejhg ARTICLE The 2q37-deletion syndrome: an update of the clinical spectrum including overweight, brachydactyly and behavioural features in 14 new patients Camille Leroy1,2,3, Emilie Landais1,2,4, Sylvain Briault5, Albert David6, Olivier Tassy7, Nicolas Gruchy8, Bruno Delobel9, Marie-Jose´ Gre´goire10, Bruno Leheup3,11, Laurence Taine12, Didier Lacombe12, Marie-Ange Delrue12, Annick Toutain13, Agathe Paubel13, Francine Mugneret14, Christel Thauvin-Robinet3,15, Ste´phanie Arpin13, Cedric Le Caignec6, Philippe Jonveaux3,10, Myle`ne Beri10, Nathalie Leporrier8, Jacques Motte16, Caroline Fiquet17,18, Olivier Brichet16, Monique Mozelle-Nivoix1,3, Pascal Sabouraud16, Nathalie Golovkine19, Nathalie Bednarek20, Dominique Gaillard1,2,3 and Martine Doco-Fenzy*,1,2,3,18 The 2q37 locus is one of the most commonly deleted subtelomeric regions. Such a deletion has been identified in 4100 patients by telomeric fluorescence in situ hybridization (FISH) analysis and, less frequently, by array-based comparative genomic hybridization (array-CGH). A recognizable ‘2q37-deletion syndrome’ or Albright’s hereditary osteodystrophy-like syndrome has been previously described. To better map the deletion and further refine this deletional syndrome, we formed a collaboration with the Association of French Language Cytogeneticists to collect 14 new intellectually deficient patients with a distal or interstitial 2q37 deletion characterized by FISH and array-CGH. Patients exhibited facial dysmorphism (13/14) and brachydactyly (10/14), associated with behavioural problems, autism or autism spectrum disorders of varying severity and overweight or obesity. The deletions in these 14 new patients measured from 2.6 to 8.8 Mb.