CO2 Mass Transfer in a Novel Photobioreactor
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DAF Truck Spare Parts and Accessories
www.europart.net DAF Truck Spare parts and accessories Spare parts to fit: XF, CF, LF Technical equipment and accessories EUROPART – Europe's No. 1 for truck, trailer, van and bus spare parts. www.europart.net Exacting standards required. Impressive quality delivered. The EUROPART brand: Excellent products, based on manufacturer standards With EUROPART own-brand products, you Our strict and careful selection of suppliers benefi t from a competitively priced alternative. refl ects our high standards. These are products Whether you need vehicle parts such as brake that impress even under extreme conditions discs, linings, batteries and fi lters or workshop and are of a quality based on manufacturer materials such as chemical products, oils and standards. This you can depend on – always. tools – our constantly growing and technically advanced range of over 6,500 own-brand products provides the right solution for every situation and every customer requirement. EUROPART – Europe's No. 1 for truck, trailer, van and bus spare parts. Dear Customers, this catalogue offers you an overview of our top sellers that are suitable for DAF trucks. From our comprehensive range we have compiled about 1,500 items for you, that are suitable for the XF, CF and LF series from model years 1997 to 2016. The catalogue is clearly divided by series in the product areas: • engine • axles and steering • lighting and electrics • drive • braking system • bodywork • suspension and damping • compressed air • cab Series overview: Model series Model year Cab variants XF Euro -

Phenomena of Perturbation in Electrical Systems
Chapter 1 Phenomena of Perturbation in Electrical Systems 1.1. Electromagnetic perturbations in energy systems 1.1.1. Introduction Power electronic systems are increasingly being used in every field; initially, they were used in the industrial sector and then used increasingly in transportation, services and housing sectors. The flexibility in the control of electrical energy explains this evolution well. For the purposes of illustration, we estimate that the electrification of service or control functions in an aircraft offers the following gains1: – 10% on the mass; – 9% onCOPYRIGHTEDfuel consumption; MATERIAL – 13% on thrust from the engines; – 15% on maintenance costs; – 10% on the buying price. 1 According to SAFRAN company, symposium SPEC 2007. 2 Electromagnetic Compatibility in Power Electronics The field of automobiles is also subject to this evolution: the development of hybrid vehicles over the last 10 years and, more recently, the re-emergence of the fully electric car (while waiting for fuel cells vehicles) are evidence of this. Already, a large number of services have been electrified in thermal engine automobiles because of the flexibility of controls (speed variation) and high yield of the electrical systems: power steering, anti-blocking system (ABS), various pumps, window winders, air conditioning (to come). The introduction of this technology, as a consequence, must take into consideration its implementation constraints; electromagnetic compatibility (EMC) in particular. Indeed, static converters based on power electronics are important sources of electromagnetic perturbations that can occasionally cause malfunctions in their local or distant electronic environment: avionics, navigation systems, reception antennae, etc. Thus, it is important to understand the origin of these phenomena, their mode of propagation and the effects on their potential “victims” in order to optimize the essential reduction or protection devices necessary to conform to the standards of EMC. -

WP7 Safety Assessment: Experimental Testing
WORK PACKAGE 7 Safety Assessment: Experimental Testing – Ignition Potential WP7 SAFETY ASSESSMENT The Hy4Heat Safety Assessment has focused on assessing the safe use of hydrogen gas in certain types of domestic properties and buildings. The evidence collected is presented in the reports listed below, all of which have been reviewed by the HSE. The summary reports (the Precis and the Safety Assessment Conclusions Report) bring together all the findings of the work and should be looked to for context by all readers. The technical reports should be read in conjunction with the summary reports. While the summary reports are made as accessible as possible for general readers, the technical reports may be most accessible for readers with a degree of technical subject matter understanding. Safety Assessment: Safety Assessment: Precis Gas Dispersion Modelling Assessment An overview of the Safety Assessment work A modelling assessment of how natural gas and undertaken as part of the Hy4Heat programme. hydrogen gas disperses and accumulates within an enclosure (e.g. in the event of a gas leak in a building). Safety Assessment: Conclusions Report Safety Assessment: (incorporating Quantitative Risk Assessment) Gas Dispersion Data Analysis A comparative risk assessment of natural gas A review of experimental data focusing on how versus hydrogen gas, including a quantitative risk natural gas and hydrogen gas disperses and assessment; and identification of control measures accumulates within an enclosure (e.g. in the event of to reduce risk and manage hydrogen gas safety for a a gas leak in a building). community demonstration. Safety Assessment: Safety Assessment: Gas Escape Frequency and Magnitude Consequence Modelling Assessment Assessment A comparative modelling assessment of the An assessment of the diferent causes of existing consequences in the event of a gas leak and ignition natural gas leaks and the frequency of such events; event for natural gas and hydrogen gas. -
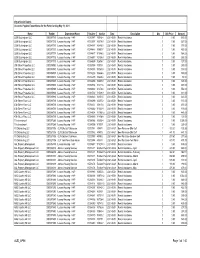
2021 05 11 Web Disb
City of Cedar Rapids Accounts Payable Expenditures for the Period ending May 11, 2021 Name Vendor Department Name Voucher Invoice Date Description Qty Unit Price Amount 2200 Buckingham LLC 0000017713 Leased Housing - HAP 00383977 V0046-4 2021-05-01 Rental Assistance 0 0.00 545.00 2200 Buckingham LLC 0000017713 Leased Housing - HAP 00383997 V0079-11 2021-05-01 Rental Assistance 0 0.00 337.00 2200 Buckingham LLC 0000017713 Leased Housing - HAP 00384187 V0486-9 2021-05-01 Rental Assistance 0 0.00 371.00 2200 Buckingham LLC 0000017713 Leased Housing - HAP 00384448 V0993-7 2021-05-01 Rental Assistance 0 0.00 465.00 2200 Buckingham LLC 0000017713 Leased Housing - HAP 00384477 V1046-2 2021-05-01 Rental Assistance 0 0.00 368.00 2200 Buckingham LLC 0000017713 Leased Housing - HAP 00384817 V2139-3 2021-05-01 Rental Assistance 0 0.00 292.00 2200 Buckingham LLC 0000017713 Leased Housing - HAP 00384899 V2604-7 2021-05-01 Rental Assistance 0 0.00 727.00 29th Street Properties LLC 0000015983 Leased Housing - HAP 00383959 V0003-1 2021-05-01 Rental Assistance 0 0.00 240.00 29th Street Properties LLC 0000015983 Leased Housing - HAP 00384035 V0163-9 2021-05-01 Rental Assistance 0 0.00 359.00 29th Street Properties LLC 0000015983 Leased Housing - HAP 00384260 V0666-6 2021-05-01 Rental Assistance 0 0.00 359.00 29th Street Properties LLC 0000015983 Leased Housing - HAP 00384275 V0685-8 2021-05-01 Rental Assistance 0 0.00 58.00 29th Street Properties LLC 0000015983 Leased Housing - HAP 00384450 V0997-11 2021-05-01 Rental Assistance 0 0.00 348.00 29th Street -

A Discrete Dimming Ballast for Linear Fluorescent Lamps
A DISCRETE DIMMING BALLAST FOR LINEAR FLUORESCENT LAMPS HAIYAN WANG Bachelor of Science in Electrical Engineering Beijing University of Aeronautics and Astronautics July, 1994 Submitted in partial fulfillment of requirement for degree MASTER OF SCIENCE OF ELECTRICAL ENGINEERING at the I CLEVELAND STATE UNIVERSITY December, 2003 CLEVELAND STATE UNIVERSITY LIBRARY OCopy-rightby ��AIYANWANG 2003 A CKNOWNLEDGEMENTS I would like to thank Dr. Ana Stankovic for the support and encouragement she gave towards the development of the thesis. Special thanks to David Kachmarik, Dr. Louis Nerone, and Timothy Chen for guiding me throughout the thesis and my time at GE lighting. I would also like to thank Dr. F.E. Villaseca and Dr. Dan Simon for their valuable suggestions for my thesis. Finally, endless thanks to my family for supporting me to gain a higher education. DISCRETE DIMMING BALLAST FOR LINEAR FLUORESCENT LAMPS HAIYAN WANG ABSTRACT A new discrete dimming ballast for the linear fluorescent lamps is proposed in this paper. The dimming control circuit is combined with a ballast module for multiple lamps to realize three discrete lighting levels control. Compared with conventional step dimming or odoff control methods, the proposed discrete dimming method has following advantages: (1) Digital signal is generated by the dimming control circuit to control the lamps on and off, which makes the system more reliable and integrated. (2) The proposed discrete dimming system replace the relays, which are necessary in conventional lamp on/off control, and decrease the system cost. (3) The proposed dimming ballast can be installed by keeping the original wiring system. This makes the upgrading lighting system more effective and efficient. -

Erres KY 159
Erres KY 159 Foto: Piet Blaas The restoration of an uncommon radio set By Jacques Hermans & Paul Bolt Index: 1. The acquisition 2. To restore or not to restore 3. The circuit diagram 4. The casing 5. First-class demolition 6. Rejuvenating the chassis 7. Repairing the electrolytic capacitors 8. The tar capacitors 9. The build-up 10. Voltage, current, music 11. Summary 1. The acquisition It is the first Saturday of august, 2008 and I am browsing a familiar exhibition for old technology in the small town Hoenderloo. Aside from some parts I had not yet made any purchases and was about to head home, when I laid my eyes on a radio set that I did not immediately recognize. The Erres-buttons instantly caught my attention. The set had endured a lot and the back wall of the casing was missing. The nameplate indicated that this was the KY 159 (serial number 1161) made in 1936 and after some negotiating the device changed hands. At home, the radio set was more thoroughly inspected. This turned out to be a letdown... Picture 1.1 The casing looked a lot less flourishing than it looked on the sunny market stand. The damage to and stains on the casing would not give in to basic cleaning and scrubbing. In some occasions furniture oil can provide a great result, which would make stripping, coloring and varnishing not necessary. Unfortunately this was not the case (picture 1.1). A quick look at the interior did not make my heart skip a beat either. -

Download the 2021 IEEE Thesaurus
2021 IEEE Thesaurus Version 1.0 Created by The Institute of Electrical and Electronics Engineers (IEEE) 2021 IEEE Thesaurus The IEEE Thesaurus is a controlled The IEEE Thesaurus also provides a vocabulary of almost 10,900 descriptive conceptual map through the use of engineering, technical and scientific terms, semantic relationships such as broader as well as IEEE-specific society terms terms (BT), narrower terms (NT), 'used for' [referred to as “descriptors” or “preferred relationships (USE/UF), and related terms terms”] .* Each descriptor included in the (RT). These semantic relationships identify thesaurus represents a single concept or theoretical connections between terms. unit of thought. The descriptors are Italic text denotes Non-preferred terms. considered the preferred terms for use in Bold text is used for preferred headings. describing IEEE content. The scope of descriptors is based on the material presented in IEEE journals, conference Abbreviations used in the Thesaurus: papers, standards, and/or IEEE organizational material. A controlled BT - Broader term vocabulary is a specific terminology used in NT - Narrower term a consistent and controlled fashion that RT - Related term results in better information searching and USE- Use preferred term retrieval. UF - Used for Thesaurus construction is based on the ANSI/NISO Z39.19-2005(2010) standard, Guidelines for the Construction, Format, and Management of Monolingual Controlled Vocabulary. The Thesaurus vocabulary uses American-based spellings with cross references to British variant spellings. The scope and structure of the IEEE Thesaurus reflects the engineering and scientific disciplines that comprise the Societies, Councils, and Communities of the IEEE in *Refer to ANSI/NISO NISO Z39.19-2005 addition to the technologies IEEE serves. -

Energy Efficiency – HID Lighting
PDHonline Course E423 (5 PDH) Energy Efficiency High Intensity Discharge Lighting Instructor: Lee Layton, P.E 2014 PDH Online | PDH Center 5272 Meadow Estates Drive Fairfax, VA 22030-6658 Phone & Fax: 703-988-0088 www.PDHonline.org www.PDHcenter.com An Approved Continuing Education Provider www.PDHcenter.com PDHonline Course E423 www.PDHonline.org Energy Efficiency High Intensity Discharge Lighting Lee Layton, P.E Table of Contents Section Page Introduction ………………………………….….. 3 Chapter 1, Lighting Market ………………….….. 5 Chapter 2, Fundamentals of Lighting ………….... 16 Chapter 3, Characteristics of HID Lighting……... 28 Chapter 4, Types of HID Lighting……………..... 37 Summary ……………………………………..…. 66 © Lee Layton. Page 2 of 66 www.PDHcenter.com PDHonline Course E423 www.PDHonline.org Introduction Gas-discharge lamps are light sources that generate light by sending an electrical discharge through an ionized gas. The character of the gas discharge depends on the pressure of the gas as well as the frequency of the current. High-intensity discharge (HID) lighting provides the highest efficacy and longest service life of any lighting type. It can save 75%-90% of lighting energy when it replaces incandescent lighting. Figure 1 shows a typical high-intensity discharge lamp. In a high-intensity discharge lamp, electricity arcs between two electrodes, creating an intensely bright light. Usually a gas of mercury, sodium, or metal halide acts as the conductor. HID lamps use an electric arc to produce intense light. Like fluorescent lamps, they require ballasts. They also take up to 10 minutes to produce light when first turned on because the ballast needs time to establish the electric arc. -

FLR) for Fusion Power
Informal Report A Conceptual Design of the Fast-Liner Reactor (FLR) for Fusion Power oCO "co LOS ALAMOS SCIENTIFIC LABORATORY Post Office Box 1663 Los Alamos. New Mexico 87545 TjyK m xmt OOCUKJE^T IS UNLKOTED LA-7686-MS Informal Report UC-20d Issued: February 1979 A Conceptual Design of the Fast-Liner Reactor (FLR) for Fusion Power R. W. Moses R. A. Krakowski R. L. Miller MAJOR CONTRIBUTORS G. E. Cort (heat transfer), R. A. Gerwin (physics), R. L. Hagenson (plasma engineering, costing), J. D. Jacobson (blast confinement), T. R. Jarboe (physics), R C. Malone (physics), T. A. Oliphant (numerical techniques), P. D. Soran (neutronics), and C. E. Swannack (energy storage and tninsfer). This work was supported by the US Department of Energy, Office of Fusion Energy, and in part by the Electric Power Research Institute. IftSl CONTENTS ABSTRACT 1 I. INTRODUCTION AND SUMMARY 1 II. SUMMARY DESCRIPTION OF REACTOR OPERATION 3 III. PHYSICS AND TECHNOLOGY DESIGN BASES 7 A. Reactor Physics 7 1. Plasma Model 7 a. Radial Transport 9 b. Axial Transport 10 c. Burn Dynamics 11 2. Liner Model 11 a. Stability 12 b. Dynamics 13 3. Numerical Methods 14 4. Development of Physics Operating Point 14 B. Reactor Engineering/Technology 20 1. Energy Balance 21 2. Plasma Preparation 23 3. Energy Storage, Switching, and Transfer 24 4. Liner Leads 26 5. Neutronics Analysis 32 6. Blast Containment 39 7. Heat Transfer 46 C. Costing Model 50 D. Design Point 52 IV. PRESENT KNOWLEDGE IN PHYSICS AND TECHNOLOGY 65 A. Physics Confidence 67 1. Plasma Preparation 67 2. -

2018 IEEE Thesaurus
2018 IEEE Thesaurus Version 1.0 Created by The Institute of Electrical and Electronics Engineers (IEEE) 2018 IEEE Thesaurus The IEEE Thesaurus is a controlled The IEEE Thesaurus also provides a vocabulary of almost 10,100 descriptive conceptual map through the use of engineering, technical and scientific terms, semantic relationships such as broader as well as IEEE-specific society terms terms (BT), narrower terms (NT), 'used for' [referred to as “descriptors” or “preferred relationships (USE/UF), and related terms terms”] .* Each descriptor included in the (RT). These semantic relationships identify thesaurus represents a single concept or theoretical connections between terms. unit of thought. The descriptors are Italic text denotes Non-preferred terms. considered the preferred terms for use in Bold text is used for preferred headings. describing IEEE content. The scope of descriptors is based on the material presented in IEEE journals, conference Abbreviations used in the Thesaurus: papers, standards, and/or IEEE organizational material. A controlled BT - Broader term vocabulary is a specific terminology used in NT - Narrower term a consistent and controlled fashion that RT - Related term results in better information searching and USE- Use preferred term retrieval. UF - Used for Thesaurus construction is based on the ANSI/NISO Z39.19-2005(2010) standard, Guidelines for the Construction, Format, and Management of Monolingual Controlled Vocabulary. The Thesaurus vocabulary uses American-based spellings with cross references to British variant spellings. The scope and structure of the IEEE Thesaurus reflects the engineering and scientific disciplines that comprise the Societies, Councils, and Communities of the IEEE in *Refer to ANSI/NISO NISO Z39.19-2005 addition to the technologies IEEE serves. -

Chapter 2 Incandescent Light Bulb
Lamp Contents 1 Lamp (electrical component) 1 1.1 Types ................................................. 1 1.2 Uses other than illumination ...................................... 2 1.3 Lamp circuit symbols ......................................... 2 1.4 See also ................................................ 2 1.5 References ............................................... 2 2 Incandescent light bulb 3 2.1 History ................................................. 3 2.1.1 Early pre-commercial research ................................ 4 2.1.2 Commercialization ...................................... 5 2.2 Tungsten bulbs ............................................. 6 2.3 Efficacy, efficiency, and environmental impact ............................ 8 2.3.1 Cost of lighting ........................................ 9 2.3.2 Measures to ban use ...................................... 9 2.3.3 Efforts to improve efficiency ................................. 9 2.4 Construction .............................................. 10 2.4.1 Gas fill ............................................ 10 2.5 Manufacturing ............................................. 11 2.6 Filament ................................................ 12 2.6.1 Coiled coil filament ...................................... 12 2.6.2 Reducing filament evaporation ................................ 12 2.6.3 Bulb blackening ........................................ 13 2.6.4 Halogen lamps ........................................ 13 2.6.5 Incandescent arc lamps .................................... 14 2.7 Electrical -

(19) United States (12) Patent Application Publication (10) Pub
111111 1111111111111111111111111111111111111111111111111111111111111111111111111111 us 20200002828Al (19) United States (12) Patent Application Publication (10) Pub. No.: US 2020/0002828 At MILLS (43) Pub. Date: Jan. 2, 2020 (54) ELECTRICAL POWER GENERATION Publication Classification SYSTEMS AND METHODS REGARDING (51) Int. Cl. SAME e25B 13104 (2006.01) H05H 1124 (2006.01) (71) Applicant: BRILLIANT LIGHT POWER, INC., HOlL 3110725 (2006.01) Cranbury, NJ (US) HOlL 3110735 (2006.01) H02S 40132 (2006.01) (72) Inventor: RANDELL L. MILLS, CRANBURY, H02S 40138 (2006.01) NJ (US) H02S 40122 (2006.01) H02S 40142 (2006.01) (73) Assignee: BRILLIANT LIGHT POWER, INC., e25B 1104 (2006.01) CRANBURY, NJ (US) (52) U.S. Cl. CPC ............... e25B 13104 (2013.01); H05H 1124 (21) Appl. No.: 16/567,689 (2013.01); HOlL 3110725 (2013.01); HOlL 3110735 (2013.01); H05H 2277/13 (2013.01); (22) Filed: Sep. 11, 2019 H02S 40138 (2014.12); H02S 40122 (2014.12); H02S 40142 (2014.12); e25B 1104 (2013.01); Related U.S. Application Data H02S 40132 (2014.12) (63) Continuation of application No. 15/314,196, filed on (57) ABSTRACT Nov. 28, 2016, now Pat. No. 10,443,139, filed as A solid or liquid fuel to plasma to electricity power source application No. PCTlUS2015/033165 on May 29, that provides at least one of electrical and thermal power 2015. comprising (i) at least one reaction cell for the catalysis of (60) Provisional application No. 62/004,883, filed on May atomic hydrogen to form hydrinos, (ii) a chemical fuel 29, 2014, provisional application No. 62/012,193, mixture comprising at least two components chosen from: a source of H 0 catalyst or H 0 catalyst; a source of atomic filed on Jun.