2. Phase Transitions 2.1 Phase Transitions in Soft Matter Soft Matter
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Phase-Transition Phenomena in Colloidal Systems with Attractive and Repulsive Particle Interactions Agienus Vrij," Marcel H
Faraday Discuss. Chem. SOC.,1990,90, 31-40 Phase-transition Phenomena in Colloidal Systems with Attractive and Repulsive Particle Interactions Agienus Vrij," Marcel H. G. M. Penders, Piet W. ROUW,Cornelis G. de Kruif, Jan K. G. Dhont, Carla Smits and Henk N. W. Lekkerkerker Van't Hof laboratory, University of Utrecht, Padualaan 8, 3584 CH Utrecht, The Netherlands We discuss certain aspects of phase transitions in colloidal systems with attractive or repulsive particle interactions. The colloidal systems studied are dispersions of spherical particles consisting of an amorphous silica core, coated with a variety of stabilizing layers, in organic solvents. The interaction may be varied from (steeply) repulsive to (deeply) attractive, by an appropri- ate choice of the stabilizing coating, the temperature and the solvent. In systems with an attractive interaction potential, a separation into two liquid- like phases which differ in concentration is observed. The location of the spinodal associated with this demining process is measured with pulse- induced critical light scattering. If the interaction potential is repulsive, crystallization is observed. The rate of formation of crystallites as a function of the concentration of the colloidal particles is studied by means of time- resolved light scattering. Colloidal systems exhibit phase transitions which are also known for molecular/ atomic systems. In systems consisting of spherical Brownian particles, liquid-liquid phase separation and crystallization may occur. Also gel and glass transitions are found. Moreover, in systems containing rod-like Brownian particles, nematic, smectic and crystalline phases are observed. A major advantage for the experimental study of phase equilibria and phase-separation kinetics in colloidal systems over molecular systems is the length- and time-scales that are involved. -

Phase Transitions in Quantum Condensed Matter
Diss. ETH No. 15104 Phase Transitions in Quantum Condensed Matter A dissertation submitted to the SWISS FEDERAL INSTITUTE OF TECHNOLOGY ZURICH¨ (ETH Zuric¨ h) for the degree of Doctor of Natural Science presented by HANS PETER BUCHLER¨ Dipl. Phys. ETH born December 5, 1973 Swiss citizien accepted on the recommendation of Prof. Dr. J. W. Blatter, examiner Prof. Dr. W. Zwerger, co-examiner PD. Dr. V. B. Geshkenbein, co-examiner 2003 Abstract In this thesis, phase transitions in superconducting metals and ultra-cold atomic gases (Bose-Einstein condensates) are studied. Both systems are examples of quantum condensed matter, where quantum effects operate on a macroscopic level. Their main characteristics are the condensation of a macroscopic number of particles into the same quantum state and their ability to sustain a particle current at a constant velocity without any driving force. Pushing these materials to extreme conditions, such as reducing their dimensionality or enhancing the interactions between the particles, thermal and quantum fluctuations start to play a crucial role and entail a rich phase diagram. It is the subject of this thesis to study some of the most intriguing phase transitions in these systems. Reducing the dimensionality of a superconductor one finds that fluctuations and disorder strongly influence the superconducting transition temperature and eventually drive a superconductor to insulator quantum phase transition. In one-dimensional wires, the fluctuations of Cooper pairs appearing below the mean-field critical temperature Tc0 define a finite resistance via the nucleation of thermally activated phase slips, removing the finite temperature phase tran- sition. Superconductivity possibly survives only at zero temperature. -

The Superconductor-Metal Quantum Phase Transition in Ultra-Narrow Wires
The superconductor-metal quantum phase transition in ultra-narrow wires Adissertationpresented by Adrian Giuseppe Del Maestro to The Department of Physics in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Physics Harvard University Cambridge, Massachusetts May 2008 c 2008 - Adrian Giuseppe Del Maestro ! All rights reserved. Thesis advisor Author Subir Sachdev Adrian Giuseppe Del Maestro The superconductor-metal quantum phase transition in ultra- narrow wires Abstract We present a complete description of a zero temperature phasetransitionbetween superconducting and diffusive metallic states in very thin wires due to a Cooper pair breaking mechanism originating from a number of possible sources. These include impurities localized to the surface of the wire, a magnetic field orientated parallel to the wire or, disorder in an unconventional superconductor. The order parameter describing pairing is strongly overdamped by its coupling toaneffectivelyinfinite bath of unpaired electrons imagined to reside in the transverse conduction channels of the wire. The dissipative critical theory thus contains current reducing fluctuations in the guise of both quantum and thermally activated phase slips. A full cross-over phase diagram is computed via an expansion in the inverse number of complex com- ponents of the superconducting order parameter (equal to oneinthephysicalcase). The fluctuation corrections to the electrical and thermal conductivities are deter- mined, and we find that the zero frequency electrical transport has a non-monotonic temperature dependence when moving from the quantum critical to low tempera- ture metallic phase, which may be consistent with recent experimental results on ultra-narrow MoGe wires. Near criticality, the ratio of the thermal to electrical con- ductivity displays a linear temperature dependence and thustheWiedemann-Franz law is obeyed. -

Density Fluctuations Across the Superfluid-Supersolid Phase Transition in a Dipolar Quantum Gas
PHYSICAL REVIEW X 11, 011037 (2021) Density Fluctuations across the Superfluid-Supersolid Phase Transition in a Dipolar Quantum Gas J. Hertkorn ,1,* J.-N. Schmidt,1,* F. Böttcher ,1 M. Guo,1 M. Schmidt,1 K. S. H. Ng,1 S. D. Graham,1 † H. P. Büchler,2 T. Langen ,1 M. Zwierlein,3 and T. Pfau1, 15. Physikalisches Institut and Center for Integrated Quantum Science and Technology, Universität Stuttgart, Pfaffenwaldring 57, 70569 Stuttgart, Germany 2Institute for Theoretical Physics III and Center for Integrated Quantum Science and Technology, Universität Stuttgart, Pfaffenwaldring 57, 70569 Stuttgart, Germany 3MIT-Harvard Center for Ultracold Atoms, Research Laboratory of Electronics, and Department of Physics, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA (Received 15 October 2020; accepted 8 January 2021; published 23 February 2021) Phase transitions share the universal feature of enhanced fluctuations near the transition point. Here, we show that density fluctuations reveal how a Bose-Einstein condensate of dipolar atoms spontaneously breaks its translation symmetry and enters the supersolid state of matter—a phase that combines superfluidity with crystalline order. We report on the first direct in situ measurement of density fluctuations across the superfluid-supersolid phase transition. This measurement allows us to introduce a general and straightforward way to extract the static structure factor, estimate the spectrum of elementary excitations, and image the dominant fluctuation patterns. We observe a strong response in the static structure factor and infer a distinct roton minimum in the dispersion relation. Furthermore, we show that the characteristic fluctuations correspond to elementary excitations such as the roton modes, which are theoretically predicted to be dominant at the quantum critical point, and that the supersolid state supports both superfluid as well as crystal phonons. -

Phase Diagrams and Phase Separation
Phase Diagrams and Phase Separation Books MF Ashby and DA Jones, Engineering Materials Vol 2, Pergamon P Haasen, Physical Metallurgy, G Strobl, The Physics of Polymers, Springer Introduction Mixing two (or more) components together can lead to new properties: Metal alloys e.g. steel, bronze, brass…. Polymers e.g. rubber toughened systems. Can either get complete mixing on the atomic/molecular level, or phase separation. Phase Diagrams allow us to map out what happens under different conditions (specifically of concentration and temperature). Free Energy of Mixing Entropy of Mixing nA atoms of A nB atoms of B AM Donald 1 Phase Diagrams Total atoms N = nA + nB Then Smix = k ln W N! = k ln nA!nb! This can be rewritten in terms of concentrations of the two types of atoms: nA/N = cA nB/N = cB and using Stirling's approximation Smix = -Nk (cAln cA + cBln cB) / kN mix S AB0.5 This is a parabolic curve. There is always a positive entropy gain on mixing (note the logarithms are negative) – so that entropic considerations alone will lead to a homogeneous mixture. The infinite slope at cA=0 and 1 means that it is very hard to remove final few impurities from a mixture. AM Donald 2 Phase Diagrams This is the situation if no molecular interactions to lead to enthalpic contribution to the free energy (this corresponds to the athermal or ideal mixing case). Enthalpic Contribution Assume a coordination number Z. Within a mean field approximation there are 2 nAA bonds of A-A type = 1/2 NcAZcA = 1/2 NZcA nBB bonds of B-B type = 1/2 NcBZcB = 1/2 NZ(1- 2 cA) and nAB bonds of A-B type = NZcA(1-cA) where the factor 1/2 comes in to avoid double counting and cB = (1-cA). -

Equation of State and Phase Transitions in the Nuclear
National Academy of Sciences of Ukraine Bogolyubov Institute for Theoretical Physics Has the rights of a manuscript Bugaev Kyrill Alekseevich UDC: 532.51; 533.77; 539.125/126; 544.586.6 Equation of State and Phase Transitions in the Nuclear and Hadronic Systems Speciality 01.04.02 - theoretical physics DISSERTATION to receive a scientific degree of the Doctor of Science in physics and mathematics arXiv:1012.3400v1 [nucl-th] 15 Dec 2010 Kiev - 2009 2 Abstract An investigation of strongly interacting matter equation of state remains one of the major tasks of modern high energy nuclear physics for almost a quarter of century. The present work is my doctor of science thesis which contains my contribution (42 works) to this field made between 1993 and 2008. Inhere I mainly discuss the common physical and mathematical features of several exactly solvable statistical models which describe the nuclear liquid-gas phase transition and the deconfinement phase transition. Luckily, in some cases it was possible to rigorously extend the solutions found in thermodynamic limit to finite volumes and to formulate the finite volume analogs of phases directly from the grand canonical partition. It turns out that finite volume (surface) of a system generates also the temporal constraints, i.e. the finite formation/decay time of possible states in this finite system. Among other results I would like to mention the calculation of upper and lower bounds for the surface entropy of physical clusters within the Hills and Dales model; evaluation of the second virial coefficient which accounts for the Lorentz contraction of the hard core repulsing potential between hadrons; inclusion of large width of heavy quark-gluon bags into statistical description. -

Thermodynamics and Kinetics of Nucleation in Binary Solutions
1 Thermodynamics and kinetics of nucleation in binary solutions Nikolay V. Alekseechkin Akhiezer Institute for Theoretical Physics, National Science Centre “Kharkov Institute of Physics and Technology”, Akademicheskaya Street 1, Kharkov 61108, Ukraine Email: [email protected] Keywords: Binary solution; phase equilibrium; binary nucleation; diffusion growth; surface layer. A new approach that is a combination of classical thermodynamics and macroscopic kinetics is offered for studying the nucleation kinetics in condensed binary solutions. The theory covers the separation of liquid and solid solutions proceeding along the nucleation mechanism, as well as liquid-solid transformations, e.g., the crystallization of molten alloys. The cases of nucleation of both unary and binary precipitates are considered. Equations of equilibrium for a critical nucleus are derived and then employed in the macroscopic equations of nucleus growth; the steady state nucleation rate is calculated with the use of these equations. The present approach can be applied to the general case of non-ideal solution; the calculations are performed on the model of regular solution within the classical nucleation theory (CNT) approximation implying the bulk properties of a nucleus and constant surface tension. The way of extending the theory beyond the CNT approximation is shown in the framework of the finite- thickness layer method. From equations of equilibrium of a surface layer with coexisting bulk phases, equations for adsorption and the dependences of surface tension on temperature, radius, and composition are derived. Surface effects on the thermodynamics and kinetics of nucleation are discussed. 1. Introduction Binary nucleation covers a wide class of processes of phase transformations which can be divided into three groups: (i) gas-liquid (or solid) transformations, (ii) liquid-gas transformations, and (iii) transformations within a condensed state. -

Suppression of Phase Separation in Lifepo4 Nanoparticles
Suppression of Phase Separation in LiFePO4 Nanoparticles During Battery Discharge Peng Bai,†,§ Daniel A. Cogswell† and Martin Z. Bazant*,†,‡ †Department of Chemical Engineering and ‡Department of Mathematics, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA §State Key Laboratory of Automotive Safety and Energy, Department of Automotive Engineering, Tsinghua University, Beijing 100084, P. R. China *Corresponding author: Dr. Martin Z. Bazant. Tel: (617)258-7039; Fax: (617)258-5766; Email: [email protected] August 8, 2011 ABSTRACT: Using a novel electrochemical phase-field model, we question the common belief that LixFePO4 nanoparticles separate into Li-rich and Li-poor phases during battery discharge. For small currents, spinodal decomposition or nucleation leads to moving phase boundaries. Above a critical current density (in the Tafel regime), the spinodal disappears, and particles fill homogeneously, which may explain the superior rate capability and long cycle life of nano-LiFePO4 cathodes. KEYWORDS: Li-ion battery, LiFePO4, phase-field model, Butler-Volmer equation, spinodal decomposition, intercalation waves. 1 Introduction. – LiFePO4-based electrochemical energy storage is one of the most promising developments for electric vehicle power systems and other high-rate applications such as power tools 2,3 and renewable energy storage. Nanoparticles of LiFePO4 have recently been used to demonstrate ultrafast battery discharge4 and high power density carbon pseudocapacitors.5 However, the -

Interval Mathematics Applied to Critical Point Transitions
Revista de Matematica:´ Teor´ıa y Aplicaciones 2005 12(1 & 2) : 29–44 cimpa – ucr – ccss issn: 1409-2433 interval mathematics applied to critical point transitions Benito A. Stradi∗ Received/Recibido: 16 Feb 2004 Abstract The determination of critical points of mixtures is important for both practical and theoretical reasons in the modeling of phase behavior, especially at high pressure. The equations that describe the behavior of complex mixtures near critical points are highly nonlinear and with multiplicity of solutions to the critical point equations. Interval arithmetic can be used to reliably locate all the critical points of a given mixture. The method also verifies the nonexistence of a critical point if a mixture of a given composition does not have one. This study uses an interval Newton/Generalized Bisection algorithm that provides a mathematical and computational guarantee that all mixture critical points are located. The technique is illustrated using several ex- ample problems. These problems involve cubic equation of state models; however, the technique is general purpose and can be applied in connection with other nonlinear problems. Keywords: Critical Points, Interval Analysis, Computational Methods. Resumen La determinaci´onde puntos cr´ıticosde mezclas es importante tanto por razones pr´acticascomo te´oricasen el modelamiento del comportamiento de fases, especial- mente a presiones altas. Las ecuaciones que describen el comportamiento de mezclas complejas cerca del punto cr´ıticoson significativamente no lineales y con multipli- cidad de soluciones para las ecuaciones del punto cr´ıtico. Aritm´eticade intervalos puede ser usada para localizar con confianza todos los puntos cr´ıticosde una mezcla dada. -

Determination of Supercooling Degree, Nucleation and Growth Rates, and Particle Size for Ice Slurry Crystallization in Vacuum
crystals Article Determination of Supercooling Degree, Nucleation and Growth Rates, and Particle Size for Ice Slurry Crystallization in Vacuum Xi Liu, Kunyu Zhuang, Shi Lin, Zheng Zhang and Xuelai Li * School of Chemical Engineering, Fuzhou University, Fuzhou 350116, China; [email protected] (X.L.); [email protected] (K.Z.); [email protected] (S.L.); [email protected] (Z.Z.) * Correspondence: [email protected] Academic Editors: Helmut Cölfen and Mei Pan Received: 7 April 2017; Accepted: 29 April 2017; Published: 5 May 2017 Abstract: Understanding the crystallization behavior of ice slurry under vacuum condition is important to the wide application of the vacuum method. In this study, we first measured the supercooling degree of the initiation of ice slurry formation under different stirring rates, cooling rates and ethylene glycol concentrations. Results indicate that the supercooling crystallization pressure difference increases with increasing cooling rate, while it decreases with increasing ethylene glycol concentration. The stirring rate has little influence on supercooling crystallization pressure difference. Second, the crystallization kinetics of ice crystals was conducted through batch cooling crystallization experiments based on the population balance equation. The equations of nucleation rate and growth rate were established in terms of power law kinetic expressions. Meanwhile, the influences of suspension density, stirring rate and supercooling degree on the process of nucleation and growth were studied. Third, the morphology of ice crystals in ice slurry was obtained using a microscopic observation system. It is found that the effect of stirring rate on ice crystal size is very small and the addition of ethylene glycoleffectively inhibits the growth of ice crystals. -

1504.05850V4.Pdf
Compact Stars in the QCD Phase Diagram IV (CSQCD IV) September 26-30, 2014, Prerow, Germany http://www.ift.uni.wroc.pl/˜csqcdiv Entropic and enthalpic phase transitions in high energy density nuclear matter Igor Iosilevskiy1,2 1 Joint Institute for High Temperatures (Russian Academy of Sciences), Izhorskaya Str. 13/2, 125412 Moscow, Russia 2 Moscow Institute of Physics and Technology (State Research University), Dolgoprudny, 141700, Moscow Region, Russia 1 Abstract Features of Gas-Liquid (GL) and Quark-Hadron (QH) phase transitions (PT) in dense nuclear matter are under discussion in comparison with their terrestrial coun- terparts, e.g. so-called ”plasma” PT in shock-compressed hydrogen, nitrogen etc. Both, GLPT and QHPT, when being represented in widely accepted temperature – baryonic chemical potential plane, are often considered as similar, i.e. amenable to one-to-one mapping by simple scaling. It is argued that this impression is illusive and that GLPT and QHPT belong to different classes: GLPT is typical enthalpic PT (Van-der-Waals-like) while QHPT (”deconfinement-driven”) is typical entropic PT. Subdivision of 1st-order fluid-fluid phase transitions into enthalpic and entropic subclasses was proposed in [arXiv:1403.8053]. Properties of enthalpic and entropic PTs differ significantly. Entropic PT is always internal part of more general and ex- tended thermodynamic anomaly – domains with abnormal (negative) sign for the set of (usually positive) second derivatives of thermodynamic potential, e.g. Gruneizen and thermal expansion and thermal pressure coefficients etc. Negative sign of these derivatives lead to violation of standard behavior and relative order for many iso-lines in P –V plane, e.g. -
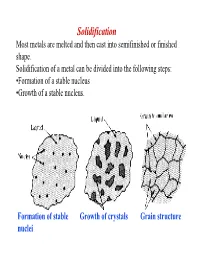
Solidification Most Metals Are Melted and Then Cast Into Semifinished Or Finished Shape
Solidification Most metals are melted and then cast into semifinished or finished shape. Solidification of a metal can be divided into the following steps: •Formation of a stable nucleus •Growth of a stable nucleus. Formation of stable Growth of crystals Grain structure nuclei Polycrystalline Metals In most cases, solidification begins from multiple sites, each of which can produce a different orientation The result is a “polycrystalline” material consisting of many small crystals of “grains” Each grain has the same crystal lattice, but the lattices are misoriented from grain to grain Driving force: solidification ⇒ For the reaction to proceed to the right ∆G AL AS V must be negative. • Writing the free energies of the solid and liquid as: S S S GV = H - TS L L L GV = H - TS ∴∴∴ ∆GV = ∆H - T∆S • At equilibrium, i.e. Tmelt , then the ∆GV = 0 , so we can estimate the melting entropy as: ∆S = ∆H/Tmelt where -∆H is the latent heat (enthalpy) of melting. • Ignore the difference in specific heat between solid and liquid, and we estimate the free energy difference as: T ∆H × ∆T ∆GV ≅ ∆H − ∆H = TMelt TMelt NUCLEATION The two main mechanisms by which nucleation of a solid particles in liquid metal occurs are homogeneous and heterogeneous nucleation. Homogeneous Nucleation Homogeneous nucleation occurs when there are no special objects inside a phase which can cause nucleation. For instance when a pure liquid metal is slowly cooled below its equilibrium freezing temperature to a sufficient degree numerous homogeneous nuclei are created by slow-moving atoms bonding together in a crystalline form.