RNA Sectors and Allosteric Function Within the Ribosome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Domain of Escherichia Coli 16S Ribosomal RNA Using Site-Directed Photoaffinity Crosslinking
Downloaded from rnajournal.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press RNA (1998), 4:1455–1466+ Cambridge University Press+ Printed in the USA+ Copyright © 1998 RNA Society+ Analysis of the conformation of the 39 major domain of Escherichia coli 16S ribosomal RNA using site-directed photoaffinity crosslinking ALEXANDRE MONTPETIT,1 CATHERINE PAYANT,1 JAMES M. NOLAN,2 and LÉA BRAKIER-GINGRAS1 1Département de Biochimie, Université de Montréal, Montréal, Québec H3T 1J4, Canada 2Department of Biochemistry, Tulane University Medical Center, New Orleans, Louisiana 70112, USA ABSTRACT The 39 major domain of Escherichia coli 16S rRNA, which occupies the head of the small ribosomal subunit, is involved in several functions of the ribosome. We have used a site-specific crosslinking procedure to gain further insights into the higher-order structure of this domain. Circularly permuted RNAs were used to introduce an azi- dophenacyl group at specific positions within the 39 major domain. Crosslinks were generated in a high-ionic strength buffer that has been used for ribosome reconstitution studies and so enables the RNA to adopt a structure recognized by ribosomal proteins. The crosslinking sites were identified by primer extension and confirmed by assessing the mobility of the crosslinked RNA lariats in denaturing polyacrylamide gels. Eight crosslinks were characterized. Among them, one crosslink demonstrates that helix 28 is proximal to the top of helix 34, and two others show that the 1337 region, located in an internal loop at the junction of helices 29, 30, 41, and 42, is proximal to the center of helix 30 and to a segment connecting helix 28 to helix 29. -

Crystal Structure of the Eukaryotic 60S Ribosomal Subunit in Complex with Initiation Factor 6
Research Collection Doctoral Thesis Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6 Author(s): Voigts-Hoffmann, Felix Publication Date: 2012 Permanent Link: https://doi.org/10.3929/ethz-a-007303759 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use. ETH Library ETH Zurich Dissertation No. 20189 Crystal Structure of the Eukaryotic 60S Ribosomal Subunit in Complex with Initiation Factor 6 A dissertation submitted to ETH ZÜRICH for the degree of Doctor of Sciences (Dr. sc. ETH Zurich) presented by Felix Voigts-Hoffmann MSc Molecular Biotechnology, Universität Heidelberg born April 11, 1981 citizen of Göttingen, Germany accepted on recommendation of Prof. Dr. Nenad Ban (Examiner) Prof. Dr. Raimund Dutzler (Co-examiner) Prof. Dr. Rudolf Glockshuber (Co-examiner) 2012 blank page ii Summary Ribosomes are large complexes of several ribosomal RNAs and dozens of proteins, which catalyze the synthesis of proteins according to the sequence encoded in messenger RNA. Over the last decade, prokaryotic ribosome structures have provided the basis for a mechanistic understanding of protein synthesis. While the core functional centers are conserved in all kingdoms, eukaryotic ribosomes are much larger than archaeal or bacterial ribosomes. Eukaryotic ribosomal rRNA and proteins contain extensions or insertions to the prokaryotic core, and many eukaryotic proteins do not have prokaryotic counterparts. Furthermore, translation regulation and ribosome biogenesis is much more complex in eukaryotes, and defects in components of the translation machinery are associated with human diseases and cancer. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -
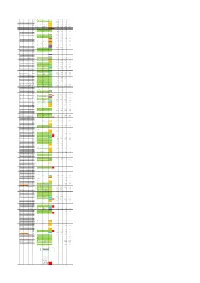
End Strand of Gene Gene Name Gene Function Starnd of Trascript
Genomic TSS strand Gene starnd of Transcipt relatively to TSS Start End Gene name coordinates relatively TSS type Control_fwd TEX_fwd Control_rev TEX_rev of gene function trascript gene group of TSS to ATG ribosomal 93 854 + rps12 + 1 protein S12 1 1 14.0638317 20.7539794 0 0 93 324 exon 1 + 111 rps12 intron 1 1 13.90756687 18.07149224 0.781323982 0.423550599 829 854 exon 2 + 496 rps12 exon2 2 24.22104343 15.24782157 0 0 30S 904 1371 + gene rps7 ribosomal + 2 protein S7 1303 -209 rps7 inter (ndhB) 2 20.47068832 9.035746118 0.625059185 0.847101199 NADH dehydrogen 1512 3535 + gene ndhB (ndh2) + ase subunit 3 2 1696 ndhB exon 1-inter 2 3.594090315 2.964854195 0.468794389 0.282367066 + 2209 ndhB exon 1-inter 2 46.09811492 4.09432246 0.468794389 0.423550599 1512 2237 exon 1 + 2756 ndhB exon 2 -inter 2 43.28534858 4.800240125 0.312529593 0.282367066 2756 3535 exon 2 + 3090 ndhB exon 2 -inter 2 17.50165719 15.95373924 0.312529593 0 + 3192 ndhB exon 2 -inter 2 140.6383167 117.6058831 2.812766334 1.694202397 - 3462 ndhB exon 2 -inter 3 1.406383167 1.129468265 1.406383167 3.67077186 4 3633 3712 + tRNA tRNA-Leu-CAA + 3610 -23 tRNA-Leu 1 77.19480938 84.85130339 0.625059185 0 + 3633 1 tRNA-Leu 1 359.5652963 649.0207016 0.781323982 0 photosyste 3954 4058 + gene psbM m II protein + 5 M 3775 -179 psbM 2 20.47068832 12.00060031 0 0.141183533 + 3954 psbM-0 2 69.22530477 28.37789015 0.156264796 0 hypothetical 4182 5073 + gene ycf66 6 protein 4182 4287 exon 1 4772 5073 exon 2 7 5202 5113 - gene ycf (ORF29) - 5299 orf29 inter 1 0 0 3.125295926 3.67077186 -

Ribosomal RNA
Ribosomal RNA Ribosomal ribonucleic acid (rRNA) is a type of non-coding RNA which is the primary component of ribosomes, essential to all cells. rRNA is a ribozyme which carries out protein synthesis in ribosomes. Ribosomal RNA is transcribed from ribosomal DNA (rDNA) and then bound to ribosomal proteins to form small and large ribosome subunits. rRNA is the physical and mechanical factor of the ribosome that forces transfer RNA (tRNA) and messenger RNA (mRNA) to process and translate the latter into proteins.[1] Ribosomal RNA Three-dimensional views of the ribosome, showing rRNA in dark blue (small subunit) is the predominant form of RNA found in most cells; it makes and dark red (large subunit). Lighter colors up about 80% of cellular RNA despite never being translated represent ribosomal proteins. into proteins itself. Ribosomes are composed of approximately 60% rRNA and 40% ribosomal proteins by mass. Contents Structure Assembly Function Subunits and associated ribosomal RNA In prokaryotes In eukaryotes Biosynthesis In eukaryotes Eukaryotic regulation In prokaryotes Prokaryotic regulation Degradation In eukaryotes In prokaryotes Sequence conservation and stability Significance Human genes See also References External links Structure Although the primary structure of rRNA sequences can vary across organisms, base-pairing within these sequences commonly forms stem-loop configurations. The length and position of these rRNA stem-loops allow them to create three-dimensional rRNA structures that are similar across species.[2] Because of these configurations, rRNA can form tight and specific interactions with ribosomal proteins to form ribosomal subunits. These ribosomal proteins contain basic residues (as opposed to acidic residues) and aromatic residues (i.e. -

Supersized Ribosomal RNA Expansion Segments in Asgard Archaea 2 Authors: Petar I
bioRxiv preprint doi: https://doi.org/10.1101/2019.12.25.888164; this version posted June 13, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Supersized ribosomal RNA expansion segments in Asgard archaea 2 Authors: Petar I. Penev1,2, Sara Fakhretaha-Aval1,3, Vaishnavi J. Patel4, Jamie J. Cannone4, 3 Robin R. Gutell4, Anton S. Petrov1,3*, Loren Dean Williams1,2,3*, Jennifer B. Glass1,2,5* 4 Affiliations: 1NASA Center for the Origin of Life, Georgia Institute of Technology, Atlanta, GA 30332-0400, USA 2School of Biological Sciences, Georgia Institute of Technology, North Avenue, Atlanta, GA 30332, USA 3School of Chemistry and Biochemistry, Georgia Institute of Technology, 901 Atlantic Dr, Atlanta, GA 30332, USA 5 4Department of Integrative Biology, The University of Texas at Austin, 2415 Speedway #C0930, 6 Austin, TX 78712, USA 7 5School of Earth and Atmospheric Sciences, Georgia Institute of Technology, 311 Ferst Dr, 8 Atlanta, GA 30332, USA 9 *Correspondence to: [email protected]; [email protected]; 10 [email protected] 11 1 bioRxiv preprint doi: https://doi.org/10.1101/2019.12.25.888164; this version posted June 13, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. -

A Systematic Rnai Synthetic Interaction Screen Reveals a Link Between P53 and Snornp Assembly
ARTICLES A systematic RNAi synthetic interaction screen reveals a link between p53 and snoRNP assembly Dragomir B. Krastev1,2, Mikolaj Slabicki2, Maciej Paszkowski-Rogacz1,2, Nina C. Hubner3,4, Magno Junqueira2,4, Andrej Shevchenko2, Matthias Mann3, Karla M. Neugebauer2 and Frank Buchholz1,2,5 TP53(tumour protein 53) is one of the most frequently mutated genes in human cancer and its role during cellular transformation has been studied extensively. However, the homeostatic functions of p53 are less well understood. Here, we explore the molecular dependency network of TP53 through an RNAi-mediated synthetic interaction screen employing two HCT116 isogenic cell lines and a genome-scale endoribonuclease-prepared short interfering RNA library. We identify a variety of TP53 synthetic interactions unmasking the complex connections of p53 to cellular physiology and growth control. Molecular dissection of the TP53 synthetic interaction with UNRIP indicates an enhanced dependency of TP53-negative cells on small nucleolar ribonucleoprotein (snoRNP) assembly. This dependency is mediated by the snoRNP chaperone gene NOLC1 (also known as NOPP140), which we identify as a physiological p53 target gene. This unanticipated function of TP53 in snoRNP assembly highlights the potential of RNAi-mediated synthetic interaction screens to dissect molecular pathways of tumour suppressor genes. More than 30 years of intense research on the tumour suppressor to different chemical drugs14,15. Several synthetic interactions with gene TP53 has revealed its relevance in many aspects of tumour tumour suppressor genes have also been published11,16, but to our biology (reviewed in ref. 1). It is now well established that p53 knowledge a genome-scale survey for synthetic interactions of a (the protein product of TP53) activation leads to the execution of a tumour suppressor gene has not been reported. -

Characterization of R-Protein Variants in Saccharomyces Cerevisiae
Characterization of r-protein variants in Saccharomyces cerevisiae DISSERTATION ZUR ERLANGUNG DES DOKTORGRADES DER NATURWISSENSCHAFTEN (DR. RER. NAT.) DER NATURWISSENSCHAFTLICHEN FAKULTÄT III BIOLOGIE UND VORKLINISCHE MEDIZIN DER UNIVERSITÄT REGENSBURG vorgelegt von Andreas Neueder aus Bogen Regensburg, 2010 Promotionsgesuch eingereicht am: 13.04.2010 Prüfungausschuss: Vorsitzender: Prof. Dr. A. Kurtz 1. Gutachter: Prof. Dr. H. Tschochner 2. Gutachter: Prof. Dr. R. Sterner 3. Prüfer: Prof. Dr. W. Seufert Ersatzprüfer: Prof. Dr. T. Dresselhaus Diese Arbeit wurde unter der Anleitung von Prof. Dr. Herbert Tschochner und Dr. Philipp Milkereit am Lehrstuhl für Biochemie III des Instituts für Mikrobiologie, Genetik und Biochemie der Universität Regensburg erstellt. Ich erkläre hiermit, dass ich die vorliegende Arbeit ohne unzulässige Hilfe Dritter und ohne Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe. Die aus anderen Quellen direkt oder indirekt übernommenen Daten und Konzepte sind unter Angabe der Quelle gekennzeichnet. Insbesondere habe ich nicht die entgeltliche Hilfe von Vermittlungs- bzw. Beratungsdiensten (Promotionsberater oder andere Personen) in Anspruch genommen. Niemand hat von mir unmittelbar oder mittelbar geldwerte Leistungen für Arbeit erhalten, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen. Die Arbeit wurde bisher weder im In- noch im Ausland in gleicher oder ähnlicher Form einer anderen Prüfungsbehörde vorgelegt. Regensburg, den Table of contents Table of contents 1 Introduction -

U·M·I University Microfilms International a Bell & Howellinformalion Company 300 North Zeeb Road, Ann Arbor, M148106-1346 USA 313/761-4700 800/521-0600
Molecular characterization of full-length cDNAs for nuclear-encoded chloroplast ribosomal proteins L12, L24 and L27 of Nicotiana tabacum. Item Type text; Dissertation-Reproduction (electronic) Authors Elhag, Gasmalla Abdelwahab. Publisher The University of Arizona. Rights Copyright © is held by the author. Digital access to this material is made possible by the University Libraries, University of Arizona. Further transmission, reproduction or presentation (such as public display or performance) of protected items is prohibited except with permission of the author. Download date 08/10/2021 23:00:38 Link to Item http://hdl.handle.net/10150/185581 INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand corner and continuing from left to right in equal sections with small overlaps. Each original is also photographed in one exposure and is included in reduced form at the back of the book. -

Jlhulm.Pdf (2.103Mb)
CHARACTERIZATION OF THE FOUR GENES ENCODING CYTOPLASMIC RIBOSOMAL PROTEIN S15a IN ARABIDOPSIS THALIANA A Thesis Submitted to the College of Graduate Studies and Research in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in the Department of Biology University of Saskatchewan Saskatoon By Jacqueline Louise Hulm © Copyright Jacqueline Louise Hulm, March, 2008. All rights reserved. PERMISSION TO USE In presenting this thesis in partial fulfillment of the requirements for a Postgraduate degree from the University of Saskatchewan, I agree that the Libraries of this University may make it freely available for inspection. I further agree that permission for copying of this thesis in any manner, in whole or in part, for scholarly purposes may be granted by the professor or professors who supervised my thesis work or, in their absence, by the Head of the Department or the Dean of the College in which my thesis work was done. It is understood that due recognition shall be given to me and to the University of Saskatchewan in any scholarly use which may be made of any material in my thesis. Requests for permission to copy or to make other use of material in this thesis in whole or in part should be addressed to: Head of the Department of Biology 112 Science Place, University of Saskatchewan Saskatoon, Saskatchewan S7N 5E2 i ABSTRACT Eukaryotic cytosolic ribosomes are composed of two distinct subunits consisting of four individual ribosomal RNAs and, in Arabidopsis thaliana, 81 ribosomal proteins. Functional subunit assembly is dependent on the equimolar production of each ribosomal component. -

Evolution of Translation the Ribosome
University of Illinois at Urbana-Champaign Luthey-Schulten Group NIH Resource for Macromolecular Modeling and Bioinformatics Computational Biophysics Workshop Evolution of Translation The Ribosome VMD Developer: John Stone MultiSeq Developers Tutorial Authors Elijah Roberts Ke Chen John Eargle John Eargle Dan Wright Tyler Earnest Jonathan Lai Zan Luthey-Schulten April 2015 A current version of this tutorial is available at http://www.scs.illinois.edu/~schulten/tutorials/ribosome CONTENTS 2 Contents Introduction 3 Requirements . 4 1 The Ribosomal SSU and associated structures: [30 minutes] 4 2 The Ribosome LSU and associated structures: [30 minutes] 9 2.1 The peptidyl-transferase center . 10 3 Ribosome Origins: [30 minutes] 11 3.1 Hypothesis on the evolution of the ribosome . 11 4 Ribosomal signatures: [60 minutes] 12 4.1 Definition and classification of the ribosomal signatures . 14 4.2 Contribution of ribosomal signatures to phylogenetic separation . 17 4.3 Functional roles of signatures in ribosomal assembly . 20 5 Kinetic Model of Ribosome assembly: [30 minutes] 22 Acknowledgements 26 CONTENTS 3 Introduction The ribosome is a large structure found in all living cells that serves as the main translation machinery of the cell. Messenger RNA (mRNA), transcribed from the organism's genome, binds with the ribosome to commence translation to protein. As explained in the previous tutorials [1, 2, 3], many other cellular components, including tRNA, the aminoacyl-tRNA synthetases, and the elonga- tion factors participate in the translation process; however, the ribosome is the central machinery that assembles a protein from a transcribed gene. Solving the structure of the ribosome was awarded the Nobel Prize in Chemistry in 2009 [4]. -

Ribosome Maturation in Yeast Models of Diamond-Blackfan Anemia and Shwachman-Diamond Syndrome
University of Louisville ThinkIR: The University of Louisville's Institutional Repository Electronic Theses and Dissertations 5-2010 Ribosome maturation in yeast models of Diamond-Blackfan anemia and Shwachman-Diamond syndrome. Joseph Brady Moore 1981- University of Louisville Follow this and additional works at: https://ir.library.louisville.edu/etd Recommended Citation Moore, Joseph Brady 1981-, "Ribosome maturation in yeast models of Diamond-Blackfan anemia and Shwachman-Diamond syndrome." (2010). Electronic Theses and Dissertations. Paper 1002. https://doi.org/10.18297/etd/1002 This Doctoral Dissertation is brought to you for free and open access by ThinkIR: The University of Louisville's Institutional Repository. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of ThinkIR: The University of Louisville's Institutional Repository. This title appears here courtesy of the author, who has retained all other copyrights. For more information, please contact [email protected]. RIBOSOME MATURATION IN YEAST MODELS OF DIAMOND-BLACKFAN ANEMIA AND SHWACHMAN-DIAMOND SYNDROME By Joseph Brady Moore IV B.S., University of Louisville, 2005 M.S., University of Louisville, 2009 A Dissertation Submitted to the Faculty of the Graduate School of the University of Louisville in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Department of Biochemistry and Molecular Biology University of Louisville Louisville, Kentucky May 2010 Copyright 2010 by Joseph B. Moore IV All rights reserved RIBOSOME MATURATION IN YEAST MODELS OF DIAMOND-BLACKFAN ANEMIA AND SHWACHMAN-DIAMOND SYNDROME By Joseph Brady Moore IV B.S., University of Louisville, 2005 M.S., University of Loiusville, 2009 A Dissertation Approved on April 12,2010 by the following Dissertation Committee: Dissertation Director: Dr.