A Systematic Rnai Synthetic Interaction Screen Reveals a Link Between P53 and Snornp Assembly
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

GEMIN8 Antibody (C-Term) Blocking Peptide Synthetic Peptide Catalog # Bp17974b
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 GEMIN8 Antibody (C-term) Blocking Peptide Synthetic peptide Catalog # BP17974b Specification GEMIN8 Antibody (C-term) Blocking GEMIN8 Antibody (C-term) Blocking Peptide - Peptide - Background Product Information The protein encoded by this gene is part of Primary Accession Q9NWZ8 the SMNcomplex, which is necessary for spliceosomal snRNP assembly in thecytoplasm and pre-mRNA splicing in the nucleus. The GEMIN8 Antibody (C-term) Blocking Peptide - Additional Information encoded proteinbinds to both SMN1 and the GEMIN6/GEMIN7 heterodimer, mediatingtheir interaction. This protein is found in nuclear Gene ID 54960 Gemini of Cajalbodies (gems) and in the cytoplasm. Three transcript variantsencoding Other Names the same protein have been found for this Gem-associated protein 8, Gemin-8, Protein gene. [providedby RefSeq]. FAM51A1, GEMIN8, FAM51A1 GEMIN8 Antibody (C-term) Blocking Format Peptides are lyophilized in a solid powder Peptide - References format. Peptides can be reconstituted in solution using the appropriate buffer as Carissimi, C., et al. J. Biol. Chem. needed. 281(48):37009-37016(2006)Carissimi, C., et al. J. Biol. Chem. 281(12):8126-8134(2006) Storage Maintain refrigerated at 2-8°C for up to 6 months. For long term storage store at -20°C. Precautions This product is for research use only. Not for use in diagnostic or therapeutic procedures. GEMIN8 Antibody (C-term) Blocking Peptide - Protein Information Name GEMIN8 Synonyms FAM51A1 Function The SMN complex plays a catalyst role in the assembly of small nuclear ribonucleoproteins (snRNPs), the building blocks of the spliceosome. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Domain of Escherichia Coli 16S Ribosomal RNA Using Site-Directed Photoaffinity Crosslinking
Downloaded from rnajournal.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press RNA (1998), 4:1455–1466+ Cambridge University Press+ Printed in the USA+ Copyright © 1998 RNA Society+ Analysis of the conformation of the 39 major domain of Escherichia coli 16S ribosomal RNA using site-directed photoaffinity crosslinking ALEXANDRE MONTPETIT,1 CATHERINE PAYANT,1 JAMES M. NOLAN,2 and LÉA BRAKIER-GINGRAS1 1Département de Biochimie, Université de Montréal, Montréal, Québec H3T 1J4, Canada 2Department of Biochemistry, Tulane University Medical Center, New Orleans, Louisiana 70112, USA ABSTRACT The 39 major domain of Escherichia coli 16S rRNA, which occupies the head of the small ribosomal subunit, is involved in several functions of the ribosome. We have used a site-specific crosslinking procedure to gain further insights into the higher-order structure of this domain. Circularly permuted RNAs were used to introduce an azi- dophenacyl group at specific positions within the 39 major domain. Crosslinks were generated in a high-ionic strength buffer that has been used for ribosome reconstitution studies and so enables the RNA to adopt a structure recognized by ribosomal proteins. The crosslinking sites were identified by primer extension and confirmed by assessing the mobility of the crosslinked RNA lariats in denaturing polyacrylamide gels. Eight crosslinks were characterized. Among them, one crosslink demonstrates that helix 28 is proximal to the top of helix 34, and two others show that the 1337 region, located in an internal loop at the junction of helices 29, 30, 41, and 42, is proximal to the center of helix 30 and to a segment connecting helix 28 to helix 29. -

Crystal Structure of the Eukaryotic 60S Ribosomal Subunit in Complex with Initiation Factor 6
Research Collection Doctoral Thesis Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6 Author(s): Voigts-Hoffmann, Felix Publication Date: 2012 Permanent Link: https://doi.org/10.3929/ethz-a-007303759 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use. ETH Library ETH Zurich Dissertation No. 20189 Crystal Structure of the Eukaryotic 60S Ribosomal Subunit in Complex with Initiation Factor 6 A dissertation submitted to ETH ZÜRICH for the degree of Doctor of Sciences (Dr. sc. ETH Zurich) presented by Felix Voigts-Hoffmann MSc Molecular Biotechnology, Universität Heidelberg born April 11, 1981 citizen of Göttingen, Germany accepted on recommendation of Prof. Dr. Nenad Ban (Examiner) Prof. Dr. Raimund Dutzler (Co-examiner) Prof. Dr. Rudolf Glockshuber (Co-examiner) 2012 blank page ii Summary Ribosomes are large complexes of several ribosomal RNAs and dozens of proteins, which catalyze the synthesis of proteins according to the sequence encoded in messenger RNA. Over the last decade, prokaryotic ribosome structures have provided the basis for a mechanistic understanding of protein synthesis. While the core functional centers are conserved in all kingdoms, eukaryotic ribosomes are much larger than archaeal or bacterial ribosomes. Eukaryotic ribosomal rRNA and proteins contain extensions or insertions to the prokaryotic core, and many eukaryotic proteins do not have prokaryotic counterparts. Furthermore, translation regulation and ribosome biogenesis is much more complex in eukaryotes, and defects in components of the translation machinery are associated with human diseases and cancer. -
![Gemin 8 (GEMIN8) Mouse Monoclonal Antibody [Clone ID: OTI4F8] Product Data](https://docslib.b-cdn.net/cover/3685/gemin-8-gemin8-mouse-monoclonal-antibody-clone-id-oti4f8-product-data-623685.webp)
Gemin 8 (GEMIN8) Mouse Monoclonal Antibody [Clone ID: OTI4F8] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA805968 Gemin 8 (GEMIN8) Mouse Monoclonal Antibody [Clone ID: OTI4F8] Product data: Product Type: Primary Antibodies Clone Name: OTI4F8 Applications: IHC, WB Recommended Dilution: WB 1:2000, IHC 1:150 Reactivity: Human Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Full length human recombinant protein of human GEMIN8 (NP_060326) produced in E.coli. Formulation: PBS (PH 7.3) containing 1% BSA, 50% glycerol and 0.02% sodium azide. Concentration: 1 mg/ml Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 28.5 kDa Gene Name: gem nuclear organelle associated protein 8 Database Link: NP_060326 Entrez Gene 54960 Human Q9NWZ8 Background: The protein encoded by this gene is part of the SMN complex, which is necessary for spliceosomal snRNP assembly in the cytoplasm and pre-mRNA splicing in the nucleus. The encoded protein binds to both SMN1 and the GEMIN6/GEMIN7 heterodimer, mediating their interaction. This protein is found in nuclear Gemini of Cajal bodies (gems) and in the cytoplasm. Three transcript variants encoding the same protein have been found for this gene. [provided by RefSeq, May 2010] This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 3 Gemin 8 (GEMIN8) Mouse Monoclonal Antibody [Clone ID: OTI4F8] – TA805968 Synonyms: FAM51A1 Product images: HEK293T cells were transfected with the pCMV6- ENTRY control (Left lane) or pCMV6-ENTRY GEMIN8 ([RC213444], Right lane) cDNA for 48 hrs and lysed. -

The Role of Nuclear Bodies in Gene Expression and Disease
Biology 2013, 2, 976-1033; doi:10.3390/biology2030976 OPEN ACCESS biology ISSN 2079-7737 www.mdpi.com/journal/biology Review The Role of Nuclear Bodies in Gene Expression and Disease Marie Morimoto and Cornelius F. Boerkoel * Department of Medical Genetics, Child & Family Research Institute, University of British Columbia, Vancouver, BC V5Z 4H4, Canada; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-604-875-2157; Fax: +1-604-875-2376. Received: 15 May 2013; in revised form: 13 June 2013 / Accepted: 20 June 2013 / Published: 9 July 2013 Abstract: This review summarizes the current understanding of the role of nuclear bodies in regulating gene expression. The compartmentalization of cellular processes, such as ribosome biogenesis, RNA processing, cellular response to stress, transcription, modification and assembly of spliceosomal snRNPs, histone gene synthesis and nuclear RNA retention, has significant implications for gene regulation. These functional nuclear domains include the nucleolus, nuclear speckle, nuclear stress body, transcription factory, Cajal body, Gemini of Cajal body, histone locus body and paraspeckle. We herein review the roles of nuclear bodies in regulating gene expression and their relation to human health and disease. Keywords: nuclear bodies; transcription; gene expression; genome organization 1. Introduction Gene expression is a multistep process that is vital for the development, adaptation and survival of all living organisms. Regulation of gene expression occurs at the level of transcription, RNA processing, RNA export, translation and protein degradation [1±3]. The nucleus has the ability to modulate gene expression at each of these levels. How the nucleus executes this regulation is gradually being dissected. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Gemin4 Is an Essential Gene in Mice, and Its Overexpression in Human Cells Causes Relocalization of the SMN Complex to the Nucleoplasm Ingo D
© 2018. Published by The Company of Biologists Ltd | Biology Open (2018) 7, bio032409. doi:10.1242/bio.032409 RESEARCH ARTICLE Gemin4 is an essential gene in mice, and its overexpression in human cells causes relocalization of the SMN complex to the nucleoplasm Ingo D. Meier1,*,§, Michael P. Walker1,2,‡,§ and A. Gregory Matera¶ ABSTRACT nuclear ribonucleoproteins (snRNPs). Each of these snRNPs Gemin4 is a member of the Survival Motor Neuron (SMN) protein contains a common set of seven RNA binding factors, called Sm complex, which is responsible for the assembly and maturation of Sm- proteins, that forms a heptameric ring around the snRNA, known as class small nuclear ribonucleoproteins (snRNPs). In metazoa, Sm the Sm core. Biogenesis of the Sm core is carried out by another snRNPs are assembled in the cytoplasm and subsequently imported macromolecular assemblage called the Survival Motor Neuron into the nucleus. We previously showed that the SMN complex is (SMN) complex, consisting of at least nine proteins (Gemins 2-8, required for snRNP import in vitro, although it remains unclear which unrip and SMN) (reviewed in Battle et al., 2006a; Matera et al., specific components direct this process. Here, we report that Gemin4 2007; Matera and Wang, 2014). overexpression drives SMN and the other Gemin proteins from the Following RNA polymerase II-mediated transcription in the cytoplasm into the nucleus. Moreover, it disrupts the subnuclear nucleus, pre-snRNAs are exported to the cytoplasm for assembly localization of the Cajal body marker protein, coilin, in a dose- into stable RNP particles (Jarmolowski et al., 1994; Ohno et al., dependent manner. -
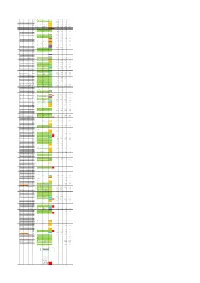
End Strand of Gene Gene Name Gene Function Starnd of Trascript
Genomic TSS strand Gene starnd of Transcipt relatively to TSS Start End Gene name coordinates relatively TSS type Control_fwd TEX_fwd Control_rev TEX_rev of gene function trascript gene group of TSS to ATG ribosomal 93 854 + rps12 + 1 protein S12 1 1 14.0638317 20.7539794 0 0 93 324 exon 1 + 111 rps12 intron 1 1 13.90756687 18.07149224 0.781323982 0.423550599 829 854 exon 2 + 496 rps12 exon2 2 24.22104343 15.24782157 0 0 30S 904 1371 + gene rps7 ribosomal + 2 protein S7 1303 -209 rps7 inter (ndhB) 2 20.47068832 9.035746118 0.625059185 0.847101199 NADH dehydrogen 1512 3535 + gene ndhB (ndh2) + ase subunit 3 2 1696 ndhB exon 1-inter 2 3.594090315 2.964854195 0.468794389 0.282367066 + 2209 ndhB exon 1-inter 2 46.09811492 4.09432246 0.468794389 0.423550599 1512 2237 exon 1 + 2756 ndhB exon 2 -inter 2 43.28534858 4.800240125 0.312529593 0.282367066 2756 3535 exon 2 + 3090 ndhB exon 2 -inter 2 17.50165719 15.95373924 0.312529593 0 + 3192 ndhB exon 2 -inter 2 140.6383167 117.6058831 2.812766334 1.694202397 - 3462 ndhB exon 2 -inter 3 1.406383167 1.129468265 1.406383167 3.67077186 4 3633 3712 + tRNA tRNA-Leu-CAA + 3610 -23 tRNA-Leu 1 77.19480938 84.85130339 0.625059185 0 + 3633 1 tRNA-Leu 1 359.5652963 649.0207016 0.781323982 0 photosyste 3954 4058 + gene psbM m II protein + 5 M 3775 -179 psbM 2 20.47068832 12.00060031 0 0.141183533 + 3954 psbM-0 2 69.22530477 28.37789015 0.156264796 0 hypothetical 4182 5073 + gene ycf66 6 protein 4182 4287 exon 1 4772 5073 exon 2 7 5202 5113 - gene ycf (ORF29) - 5299 orf29 inter 1 0 0 3.125295926 3.67077186 -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Ribosomal RNA
Ribosomal RNA Ribosomal ribonucleic acid (rRNA) is a type of non-coding RNA which is the primary component of ribosomes, essential to all cells. rRNA is a ribozyme which carries out protein synthesis in ribosomes. Ribosomal RNA is transcribed from ribosomal DNA (rDNA) and then bound to ribosomal proteins to form small and large ribosome subunits. rRNA is the physical and mechanical factor of the ribosome that forces transfer RNA (tRNA) and messenger RNA (mRNA) to process and translate the latter into proteins.[1] Ribosomal RNA Three-dimensional views of the ribosome, showing rRNA in dark blue (small subunit) is the predominant form of RNA found in most cells; it makes and dark red (large subunit). Lighter colors up about 80% of cellular RNA despite never being translated represent ribosomal proteins. into proteins itself. Ribosomes are composed of approximately 60% rRNA and 40% ribosomal proteins by mass. Contents Structure Assembly Function Subunits and associated ribosomal RNA In prokaryotes In eukaryotes Biosynthesis In eukaryotes Eukaryotic regulation In prokaryotes Prokaryotic regulation Degradation In eukaryotes In prokaryotes Sequence conservation and stability Significance Human genes See also References External links Structure Although the primary structure of rRNA sequences can vary across organisms, base-pairing within these sequences commonly forms stem-loop configurations. The length and position of these rRNA stem-loops allow them to create three-dimensional rRNA structures that are similar across species.[2] Because of these configurations, rRNA can form tight and specific interactions with ribosomal proteins to form ribosomal subunits. These ribosomal proteins contain basic residues (as opposed to acidic residues) and aromatic residues (i.e. -

Supersized Ribosomal RNA Expansion Segments in Asgard Archaea 2 Authors: Petar I
bioRxiv preprint doi: https://doi.org/10.1101/2019.12.25.888164; this version posted June 13, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Supersized ribosomal RNA expansion segments in Asgard archaea 2 Authors: Petar I. Penev1,2, Sara Fakhretaha-Aval1,3, Vaishnavi J. Patel4, Jamie J. Cannone4, 3 Robin R. Gutell4, Anton S. Petrov1,3*, Loren Dean Williams1,2,3*, Jennifer B. Glass1,2,5* 4 Affiliations: 1NASA Center for the Origin of Life, Georgia Institute of Technology, Atlanta, GA 30332-0400, USA 2School of Biological Sciences, Georgia Institute of Technology, North Avenue, Atlanta, GA 30332, USA 3School of Chemistry and Biochemistry, Georgia Institute of Technology, 901 Atlantic Dr, Atlanta, GA 30332, USA 5 4Department of Integrative Biology, The University of Texas at Austin, 2415 Speedway #C0930, 6 Austin, TX 78712, USA 7 5School of Earth and Atmospheric Sciences, Georgia Institute of Technology, 311 Ferst Dr, 8 Atlanta, GA 30332, USA 9 *Correspondence to: [email protected]; [email protected]; 10 [email protected] 11 1 bioRxiv preprint doi: https://doi.org/10.1101/2019.12.25.888164; this version posted June 13, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license.