Transgenerational Epigenetic Inheritance of the Stress Response in Rats
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

As of May 16, 2021 Member List
As Of May 16, 2021 Member List First Middle Last Designation Website Clarice Nell Aaron Genevieve Abate http://genabate333.fineartstudioonline.com Lynn Abbott http://www.lynnabbottstudios.com Kim Abernethy http://www.kimabernethy.com Liz Abeyta http://www.lizabeyta.com Bryan Abing Susan Abma http://www.susanabma.com Sharon Abshagen http://www.sharonabshagenart.com Angela Marie Accarino http://www.sunfishart.com Benone Achiriloaie Sandra Ackerman Ed Acuna Amy Adams http://www.Amyadamsfineart.com Michael Lynn Adams http://www.MichaelLynnAdams.com Peter Adams OPAM http://www.Americanlegacyfinearts.com Warren W. Adams http://www.wyomingartist.net Dustin Adamson http://www.dustinadamsonfineart.com Robert N. Adamson OPA Teresa Adaszynska http://www.adaszynskafineart.com Cyrus Afsary OPAM Priya Ahlawat http://www.priyaahlawat.com Susan Aitcheson Michael Aitken Robert T. Akers http://www.robertakers.com Robert Lawrence Akey http://www.robakey.com Daud Akhriev OPAM http://www.daudakhriev.com Lee Alban OPA http://www.leealban.com Ann H. Aldinger https://www.annaldinger.com Christy Carroll Aldredge http://www.christyaldredge.com Edward Aldrich OPA http://www.edwardaldrich.com Anne Aleshire http://www.annealeshire.com Ann Alexander Charles David Alexander http://www.charlesdavidalexander.com Gloria Alexander Joseph Alexander http://www.josephalexanderstudio.com Richard Alexander http://www.richalexanderart.com Stanton deForest Allaben http://www.stantonallabenart.com Martha Allan Laverty http://www.marthalaverty.com Laura Lawson Allee http://www.llawsonallee.faso.com/ -
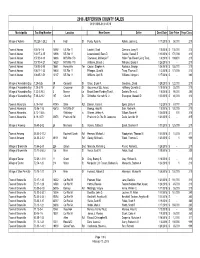
2018 Sales Report
2018 JEFFERSON COUNTY SALES 01/01/2018-01/31/2018 Municipality Tax Map NumberLocation New Owner Seller Deed Date Sale Price Prop Class Village of Adams 112.28-1-28.2 15 High St Purdy, Ryan A. Rohde, Joanne L 1/17/2018$ 58,000 210 Town of Adams 100.14-1-6 14592 US Rte 11 Laisdell, Scott Clemons, Larry R 1/8/2018$ 135,000 210 Town of Adams 100.17-2-15 13906 US Rte 11 Lewandowski, Sean D. Gaulke, Russell S 1/10/2018$ 170,000 210 Town of Adams 107.00-4-13 16686 NYS Rte 178 Townsend, McKenzie F. Haller Fam Revoc Living Trust, 1/8/2018$ 169,600 210 Town of Adams 107.00-4-21 16520 NYS Rte 178 Williams, Brandi J. Williams, Brandi J 1/26/2018$ - 210 Town of Adams 108.05-2-80 18661 Honeyville Ter Casion, Stephen A. Roukous, George 1/26/2018$ 155,000 210 Town of Adams 108.17-1-33 10604 US Rte 11 Pfleegor, Chad M. Tharp, Thomas S 1/3/2018$ 173,000 210 Town of Adams 108.05-1-51 13137 US Rte 11 Williams, April R. Williams, Morgan J 1/17/2018$ - 280 Village of Alexandria Bay 7.29-3-26 39 Cornwall St Olson, Bryan R. Goudreau, David 1/29/2018$ 122,000 210 Village of Alexandria Bay 7.29-3-78 81 Crossmon St Gouverneur S&L Assoc, Hoffberg, Camette C 1/18/2018$ 25,000 210 Village of Alexandria Bay 7.22-2-29.3 3 Mance Ln Broad Street Funding Trust I, Daniels, Emma L 1/5/2018$ 95,000 280 Village of Alexandria Bay 7.38-2-21.2 107 Church St Schreiber, Kenneth M. -

Iveigh Ivelan Iveland Ivelane Ivelant Ivelen Ivelend Ivelent Ivelind Ivelint
Iveigh Ivones Izearde Izzach Izzox Ivelan Ivoomb Izeart Izzack Izzoyd Iveland Ivoombe Izech Izzacke Izzsard Ivelane Ivor Izeck Izzaech Izzude Ivelant Ivors Izerd Izzaeck Izzume Ivelen Ivory Izert Izzaick Izzyck Ivelend Ivvaren Izett Izzaitch Izzyke Ivelent Ivy Iziac Izzake Ivelind Ivye Izick Izzaock Ivelint Ivymey Izitch Izzaox Ivelyn Ivys Izock Izzard Ivelynd Iwadby Izod Izzarde Ivemey Iwardbey Izode Izzarn Ivemmy Iwardby Izoed Izzarne Ivemy Iwardy Izoit Izzart Iveney Iwerdby Izold Izzarte Ivens Iwoodby Izombe Izzat Iver Ix Izon Izzatt Iverach Ixton Izood Izzayck Iverack Iyon Izoode Izzayke Iveragh Iyone Izoold Izzeard Iverake Iyonne Izoomb Izzearde Iverech Izaac Izoombe Izzeart Ivereck Izaach Izord Izzech Iverick Izaack Izould Izzeck Iveritch Izaacs Izoyd Izzerd Iverock Izaacson Izrael Izzert Iverox Izaake Izraeler Izzett Ivers Izach Izraely Izzick Iverson Izack Izreeli Izzitch Ivery Izacke Izsard Izzock Iveryck Izaech Izsarde Izzod Iveryke Izaeck Izsardt Izzode Ives Izaick Izsart Izzoed Iveson Izaitch Izsarte Izzoit Ivet Izake Izseard Izzold Ivett Izaock Izsearde Izzolm Ivetts Izaox Izseart Izzomb Ivey Izard Izserd Izzombe Ivie Izarde Izsert Izzome Ivimey Izarn Izsord Izzone Ivinges Izarne Izude Izzood Ivington Izart Izyck Izzoode Ivinney Izarte Izyke Izzoold Ivins Izat Izzaac Izzoom Ivinson Izatson Izzaach Izzoomb Iviormy Izatt Izzaack Izzoombe Ivison Izayck Izzaacs Izzord Ivombe Izayke Izzaacson Izzould Ivone Izeard Izzaake Izzown Hall of Names by Swyrich © 1999 Swyrich Corporation www.swyrich.com 1-888-468-7686 557 Jackline Jacok -

Endometrial Response to Conceptus-Derived Estrogen and Interleukin-1Β at the Time of Implantation in Pigs
Ka et al. Journal of Animal Science and Biotechnology (2018) 9:44 https://doi.org/10.1186/s40104-018-0259-8 REVIEW Open Access Endometrial response to conceptus- derived estrogen and interleukin-1β at the time of implantation in pigs Hakhyun Ka1*, Heewon Seo1,2, Yohan Choi1,3, Inkyu Yoo1 and Jisoo Han1 Abstract: The establishment of pregnancy is a complex process that requires a well-coordinated interaction between the implanting conceptus and the maternal uterus. In pigs, the conceptus undergoes dramatic morphological and functional changes at the time of implantation and introduces various factors, including estrogens and cytokines, interleukin-1β2 (IL1B2), interferon-γ (IFNG), and IFN-δ (IFND), into the uterine lumen. In response to ovarian steroid hormones and conceptus-derived factors, the uterine endometrium becomes receptive to the implanting conceptus by changing its expression of cell adhesion molecules, secretory activity, and immune response. Conceptus-derived estrogens act as a signal for maternal recognition of pregnancy by changing the direction of prostaglandin (PG) F2α from the uterine vasculature to the uterine lumen. Estrogens also induce the expression of many endometrial genes, including genes related to growth factors, the synthesis and transport of PGs, and immunity. IL1B2, a pro-inflammatory cytokine, is produced by the elongating conceptus. The direct effect of IL1B2 on endometrial function is not fully understood. IL1B activates the expression of endometrial genes, including the genes involved in IL1B signaling and PG synthesis and transport. In addition, estrogen or IL1B stimulates endometrial expression of IFN signaling molecules, suggesting that estrogen and IL1B act cooperatively in priming the endometrial function of conceptus-produced IFNG and IFND that, in turn, modulate endometrial immune response during early pregnancy. -

Okhrana Records
http://oac.cdlib.org/findaid/ark:/13030/kt538nf189 Online items available Register of the Okhrana records Finding aid prepared by Andrej Kobal and Sally DeBauche Hoover Institution Archives 434 Galvez Mall Stanford University Stanford, CA, 94305-6010 (650) 723-3563 [email protected] © 1964, 2016 Register of the Okhrana records 26001 1 Title: Okhrana records Date (inclusive): 1883-1917 Collection Number: 26001 Contributing Institution: Hoover Institution Archives Language of Material: Russian Physical Description: 232 manuscript boxes, 86 card file boxes, 6 oversize boxes(194.6 linear feet) Abstract: Intelligence reports from agents in the field and the Paris office, dispatches, circulars, headquarters studies, correspondence of revolutionaries, and photographs, relating to activities of Russian revolutionists abroad. Collection is available on microfilm (509 reels). Digital copies of select records also available at https://digitalcollections.hoover.org. Physical Location: Hoover Institution Archives Creator: Russia. Departament politsii. Zagranichnaia agentura (Paris) Access Microfilm use only. Digital copies of select records also available at https://digitalcollections.hoover.org. The Hoover Institution Archives only allows access to copies of audiovisual items. To listen to sound recordings or to view videos or films during your visit, please contact the Archives at least two working days before your arrival. We will then advise you of the accessibility of the material you wish to see or hear. Please note that not all audiovisual material is immediately accessible. Publication Rights For copyright status, please contact the Hoover Institution Archives. Preferred Citation [Identification of item], Okhrana records, [Index number, Folder number], Hoover Institution Archives. Acquisition Information Acquired by the Hoover Institution Archives in 1926. -

Urine-Derived Epithelial Cells As Models for Genetic Kidney Diseases
cells Review Urine-Derived Epithelial Cells as Models for Genetic Kidney Diseases Tjessa Bondue 1 , Fanny O. Arcolino 1 , Koenraad R. P. Veys 1,2, Oyindamola C. Adebayo 1,3, Elena Levtchenko 1,2, Lambertus P. van den Heuvel 1,4 and Mohamed A. Elmonem 5,* 1 Department of Development and Regeneration, KU Leuven, 3000 Leuven, Belgium; [email protected] (T.B.); [email protected] (F.O.A.); [email protected] (K.R.P.V.); [email protected] (O.C.A.); [email protected] (E.L.); [email protected] (L.P.v.d.H.) 2 Department of Pediatrics, Division of Pediatric Nephrology, University Hospitals Leuven, 3000 Leuven, Belgium 3 Centre for Molecular and Vascular Biology, Department of Cardiovascular Sciences, KU Leuven, 3000 Leuven, Belgium 4 Department of Pediatric Nephrology, Radboud University Medical Center, 6500 Nijmegen, The Netherlands 5 Department of Clinical and Chemical Pathology, Faculty of Medicine, Cairo University, Cairo 11628, Egypt * Correspondence: [email protected] Abstract: Epithelial cells exfoliated in human urine can include cells anywhere from the urinary tract and kidneys; however, podocytes and proximal tubular epithelial cells (PTECs) are by far the most relevant cell types for the study of genetic kidney diseases. When maintained in vitro, they have been proven extremely valuable for discovering disease mechanisms and for the development of new therapies. Furthermore, cultured patient cells can individually represent their human sources and their specific variants for personalized medicine studies, which are recently gaining much Citation: Bondue, T.; Arcolino, F.O.; interest. In this review, we summarize the methodology for establishing human podocyte and PTEC Veys, K.R.P.; Adebayo, O.C.; cell lines from urine and highlight their importance as kidney disease cell models. -

The Effect of Abstinence from Smoking on Stress Reactivity" (2019)
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2019 The ffecE t Of Abstinence From Smoking On Stress Reactivity Cheyenne Allenby University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Pharmacology Commons Recommended Citation Allenby, Cheyenne, "The Effect Of Abstinence From Smoking On Stress Reactivity" (2019). Publicly Accessible Penn Dissertations. 3295. https://repository.upenn.edu/edissertations/3295 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/3295 For more information, please contact [email protected]. The ffecE t Of Abstinence From Smoking On Stress Reactivity Abstract Subjective stress is a well-documented predictor of early smoking relapse, yet our understanding of stress and tobacco use is limited by the reliability of current available measures of stress. Functional magnetic reasoning imaging (fMRI) could provide a much-needed objective measure of stress reactivity. The og al of this dissertation is to contribute to the understanding of abstinence-induced changes in stress reactivity by examining neural, neuroendocrine (cortisol), and subjective measures of stress response during abstinence. In addition, this study investigated the influence of individual variation in nicotine metabolism rates on these measures of stress reactivity. Seventy-five treatment-seeking smokers underwent blood oxygen level dependent (BOLD) fMRI during the Montreal Imaging Stress Task (MIST) on two occasions: once during smoking satiety and once following biochemically confirmed 24-hour abstinence (order counter-balanced). The primary outcome measure was brain response during stress (vs. control) blocks of the MIST. Neural stress reactivity during abstinence (vs. satiety) was associated with significantly increased activation in the left inferior frontal gyrus (IFG), a brain region previously associated with inhibitory control. -

ATP Rankings
ATP Rankings Rankings Date: Sep 27, 2021 Total Grand Slam Masters Other Tourns. Points Next Rank # Player Natl. Points Points 1000 Points Points Played Dropping Best 1 Djokovic, Novak SRB 12133 7200 2438 2495 18 500 0 2 Medvedev, Daniil RUS 10575 3740 3810 3025 26 0 0 3 Tsitsipas, Stefanos GRE 8350 2020 2920 3410 29 300 125 4 Zverev, Alexander GER 7760 2340 3560 1860 27 180 23 5 Rublev, Andrey RUS 6130 810 1830 3490 30 0 0 6 Nadal, Rafael ESP 5815 1720 2400 1695 16 0 0 7 Berrettini, Matteo ITA 5173 2100 1150 1923 24 0 0 8 Thiem, Dominic AUT 4995 1385 1360 2250 21 500 0 9 Federer, Roger SUI 3765 1500 1070 1195 14 0 0 10 Ruud, Casper NOR 3440 325 1295 1820 35 0 0 11 Auger-Aliassime, Felix CAN 3368 1270 503 1595 32 0 0 12 Hurkacz, Hubert POL 3333 820 1563 950 33 0 0 13 Shapovalov, Denis CAN 3265 923 1050 1292 33 0 0 14 Sinner, Jannik ITA 2895 415 780 1700 41 250 40 15 Schwartzman, Diego ARG 2800 810 760 1230 28 45 23 16 Carreno Busta, Pablo ESP 2550 290 565 1695 28 0 0 17 Garin, Cristian CHI 2510 450 540 1520 34 0 0 18 Bautista Agut, Roberto ESP 2360 585 865 910 29 0 0 19 Opelka, Reilly USA 2341 360 1116 865 31 180 0 20 Monfils, Gael FRA 2278 360 640 1278 26 0 0 21 Isner, John USA 2171 213 1075 883 22 90 10 22 Evans, Daniel GBR 2164 325 485 1354 37 65 23 23 Sonego, Lorenzo ITA 2097 325 698 1074 38 0 0 24 Karatsev, Aslan RUS 2072 890 290 892 43 32 10 25 Humbert, Ugo FRA 2045 155 315 1575 41 0 0 26 de Minaur, Alex AUS 1991 168 235 1588 31 45 10 27 Khachanov, Karen RUS 1965 550 505 910 33 180 45 28 Norrie, Cameron GBR 1930 280 230 1420 33 65 -

Panel Addresses Racism at ND
VOL. XXV. NO. 94 The ObserverWEDNESDAY, FEBRUARY 17, 1993 THE INDEPENDENT NEWSPAPER SERVING NOTRE DAME AND SAINT MARY'S Panel addresses racism at ND • See insert on Race residence halls at Notre Dame. By CAROLINE CLARKE An hispanic student who was News Writer Relations at Notre Dame cooking tacos and enchiladas on cultural mversny for Notre was harassed by a hall rector, Racism is a powerful, nega Dame professors intended to who found the smell tive force, and it feeds on big address racial problems within "offensive." otry, prejudice and feelings of the community In another incident, when a insecurity, according to Angela She said she saw two ex piece of lounge furniture was Borelli. tremes. found missing, the rector initi St. Edward's Hall hosted an The first was the professor ated a search of the rooms of open forum with a panel of who was "pure of heart." He minority students, said Borelli. three speakers b~gan the fancied himself as a knight in Such examples show that evening by sharing racial inci shining armor pledging to fight racism is not limited to making dents and personal anecdotes all racism, according to Borelli. slurs and physical confronta reflecting on their experiences The other extreme was the tions, according to Borelli. at the University. professor who was concerned Smith likened Notre Dame's Borelli, Assistant Professional about the minorities who at campus to a beautiful garden Specialist of Romance Lan tended his classes. that it rests upon an old battle guage and Literature, was Some professors claimed that field riddled with latent mine joined by Roland Smith, the minority students were aca fields. -

Srf Return Provider List
SRF RETURN PROVIDER LIST REF # LAST NAME CITY R_2309-1 Armstrong Andrew C., O.D. C/O Merrimack Valley Counseling Center Nashua R_2377-1 Armstrong Nancy M. , O.D. Campton R_2377-2 Armstrong Nancy M. , O.D. Plymouth R_2377-3 Armstrong Nancy M. , O.D. C/O Women's Medical Assoc Of Nashua R_566-3 Aronson Mark J. , D.O. Nashua R_566-4 Aronson Mark J. , D.O. Laconia R_566-1 Aronson Mark J. , D.O. C/O Mitchell B Jean Esq Gilford R_566-2 Aronson Mark J. , D.O. C/O Mitchell B Jean Esq Laconia R_1369 Arsenault Brian J. , DC Pelham R_1267 Arsenault Diane Louise , M.D. Mid-State Health Center Plymouth R_1772 Arsenault Joseph B. , DC Pelham R_2703 Arsenault Raymond E. Salem R_2645 Arvidson Dwight B. OD C/O Judson Dexter Keene R_1173 Ashok Shanthamallappa , MD., c/o Granite State Anesthesiologists PA Nashua R_174 Asplund Charles M. , MD Muskegon R_55 Associated Anesthesiologists Of Manchester PA Manchester R_728 Associated Anesthesiologists Of Manchester PA Manchester R_37-1 Associated Radiologists, P.A. Nashua R_37-2 Associated Radiologists, P.A. Nashua R_1079 Associates In Medicine Of Valley Regional Hospital Meriden R_664 Astarjian Henry D M.D.Indiv &/Or Nashua Neurological Center PC Hollis R_2028 Astolfi Thomas M. , MD., Attn Jeannine V Forand,C/O Matthew Thornton Hlth Plan Nashua R_697 Astolfi Thomas M. Barton, M.D., Barton Orthopaedics Pllc Wolfeboro R_1656-1 Astorian Donald , M.D. Granville R_473 A. Sheinbaum MD, J. Meadows M.D.&/Or Concord Gastroenterology Concord R_1617 Abbott Robert C / Re: Paul F Racicot, M.D. Laconia R_2139 Abbott Robert C / Re: Scott Hobson MD, Lakes Region General Hospital Laconia R_305 Abbott Robert C / Re: Brian J. -

S M Ith Son Ian in Stitu Tion / 2 0 15 a Nnu a L R E P
Now+Next Smithsonian Institution / 2015 Annual Report The National Museum of African American History and Culture will open to the public Sept. 24, 2016. table of contents 2 Secretary’s Letter photo 4 2015 Milestones 6 Art Now+Next 14 Science Now+Next 22 History & Culture Now+Next 30 Education & Outreach Now+Next 40 Donors to the Smithsonian Wynton Marsalis performs at the Installation ceremony for 13th Secretary of the Smithsonian David J. Skorton. Now+Next Few events in my life have been as uplifting and humbling as that of Oct. 19, 2015, the day I was honored to become the 13th Secretary of the Smithsonian Institution. The Installation ceremony began with a procession of the institution’s scholars, curators and scientists, representing not only the more than 6,000 staff who dedicate themselves to the Smithsonian’s mission today, but also the many thousands more who have advanced the fields of history, art and science since the institution’s founding in 1846. In 2015 alone we welcomed hundreds of thousands of people to the newly renovated Renwick Gallery (p. 6), our scientists peered to the edge of the black hole at the center of the Milky Way (p. 18) and our anthropologists helped identify the remains of leaders of the Jamestown colony (p. 28). The theme of this year’s annual report is “Now+Next” because the work we do today leads to the innovations of tomorrow. Thanks to the support of many generous private donations, Congress and the American people, we will continue to increase our understanding of the world, and to share that understanding with you. -

PETITION List 05-15-13 Columns
PETITION TO FREE LYNNE STEWART: SAVE HER LIFE – RELEASE HER NOW! • 1 Signatories as of 05/15/13 Arian A., Brooklyn, New York Diane Abell, Patchogue, New York Clare A., Redondo Beach, California Emily Abendroth, Philadelphia, Pennsylvania K. A., Mexico Warren Abercrombie, Pelzer, South Carolina Kassim S. A., Malaysia Sam Abercromby, Vianna do Castello, Portugal N. A., Philadelphia, Pennsylvania Teresa Ableiter, Saint Paul, Minnesota Tammi A., Jamaica Plain, Massachusetts Sharon Abner, Washington Depot, Connecticut Tristan A., Fort Mill, South Carolina Noerena Abookire, PhD., Santa Monica, California Cory a'Ghobhainn, Los Angeles, California James Abourezk, Sioux Falls, South Dakota Tajwar Aamir, Lawrenceville, New Jersey Nardos Abraha, Hamilton, Canada Aaron Aarons, Miami, Florida Judy Abraham, London, United Kingdom Rashid Abass, Malabar, Port Elizabeth, South Kelvin Abraham, London, United Kingdom Africa Mercedes Abraham, San Francisco, California Jamshed Abbas, Vancouver, Canada Peter Abrahamson, Australia Mansoor Abbas, Southington, Connecticut Ilana Abramovitch, Brooklyn, New York Andrew Abbey, Pleasanton, California Alexis Abrams, Los Angeles, California Andrea Abbott, Oceanport, New Jersey Danielle Abrams, Ann Arbor, Michigan Clement Abbott, jackson, Georgia Danielle Abrams, Brooklyn, New York Laura Abbott, Woodstock, New York Geoffrey Abrams, New York, New York Youssef Abdalla, Round Rock, Texas Susan Abrams, West Linn, Oregon Asad Abdallah, Houston, Texas Tami Abrams, Oakland, California Samiha Abdeldjebar, Corsham, United Kingdom