The Effect of Abstinence from Smoking on Stress Reactivity" (2019)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gabab Regulation of Methamphetamine-Induced Associative Learning
Loyola University Chicago Loyola eCommons Dissertations Theses and Dissertations 2010 Gabab Regulation of Methamphetamine-Induced Associative Learning Robin Michelle Voigt Loyola University Chicago Follow this and additional works at: https://ecommons.luc.edu/luc_diss Part of the Pharmacology Commons Recommended Citation Voigt, Robin Michelle, "Gabab Regulation of Methamphetamine-Induced Associative Learning" (2010). Dissertations. 38. https://ecommons.luc.edu/luc_diss/38 This Dissertation is brought to you for free and open access by the Theses and Dissertations at Loyola eCommons. It has been accepted for inclusion in Dissertations by an authorized administrator of Loyola eCommons. For more information, please contact [email protected]. This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 3.0 License. Copyright © 2010 Robin Michelle Voigt LOYOLA UNIVERSITY CHICAGO GABAB REGULATION OF METHAMPHETAMINE-INDUCED ASSOCIATIVE LEARNING A DISSERTATION SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL IN CANDIDACY FOR THE DEGREE OF DOCTOR OF PHILOSOPHY PROGRAM IN MOLECULAR PHARMACOLOGY & THERAPEUTICS BY ROBIN MICHELLE VOIGT CHICAGO, IL DECEMBER 2010 Copyright by Robin Michelle Voigt, 2010 All rights reserved ACKNOWLEDGEMENTS Without the support of so many generous and wonderful individuals I would not have been able to be where I am today. First, I would like to thank my Mother for her belief that I could accomplish anything that I set my mind to. I would also like to thank my dissertation advisor, Dr. Celeste Napier, for encouraging and challenging me to be better than I thought possible. I extend gratitude to my committee members, Drs. Julie Kauer, Adriano Marchese, Micky Marinelli, and Karie Scrogin for their guidance and insightful input. -

As of May 16, 2021 Member List
As Of May 16, 2021 Member List First Middle Last Designation Website Clarice Nell Aaron Genevieve Abate http://genabate333.fineartstudioonline.com Lynn Abbott http://www.lynnabbottstudios.com Kim Abernethy http://www.kimabernethy.com Liz Abeyta http://www.lizabeyta.com Bryan Abing Susan Abma http://www.susanabma.com Sharon Abshagen http://www.sharonabshagenart.com Angela Marie Accarino http://www.sunfishart.com Benone Achiriloaie Sandra Ackerman Ed Acuna Amy Adams http://www.Amyadamsfineart.com Michael Lynn Adams http://www.MichaelLynnAdams.com Peter Adams OPAM http://www.Americanlegacyfinearts.com Warren W. Adams http://www.wyomingartist.net Dustin Adamson http://www.dustinadamsonfineart.com Robert N. Adamson OPA Teresa Adaszynska http://www.adaszynskafineart.com Cyrus Afsary OPAM Priya Ahlawat http://www.priyaahlawat.com Susan Aitcheson Michael Aitken Robert T. Akers http://www.robertakers.com Robert Lawrence Akey http://www.robakey.com Daud Akhriev OPAM http://www.daudakhriev.com Lee Alban OPA http://www.leealban.com Ann H. Aldinger https://www.annaldinger.com Christy Carroll Aldredge http://www.christyaldredge.com Edward Aldrich OPA http://www.edwardaldrich.com Anne Aleshire http://www.annealeshire.com Ann Alexander Charles David Alexander http://www.charlesdavidalexander.com Gloria Alexander Joseph Alexander http://www.josephalexanderstudio.com Richard Alexander http://www.richalexanderart.com Stanton deForest Allaben http://www.stantonallabenart.com Martha Allan Laverty http://www.marthalaverty.com Laura Lawson Allee http://www.llawsonallee.faso.com/ -

Effect of a Brief Memory Updating Intervention On
1 1 2 PI Name: Michael E. Saladin, PhD 3 4 Study Title: Reducing Smoking Cue Reactivity and Behavior via Retrieval-Extinction Mechanism 5 A. Specific Aims 6 The goal of the study will be to adopt many of the procedures and parameters of retrieval- 7 extinction training in an attempt to demonstrate enduring reductions in craving and cue-reactivity 8 among cigarette smokers. The study will conduct an exploratory analysis of the intervention’s effects on 9 substance use (i.e., smoking behavior). This exploratory study will investigate a novel behavioral 10 strategy for altering important memory processes that underlie human smoking-related nicotine 11 addiction. This strategy represents a paradigm shift in that it employs established cue exposure 12 procedures to putatively update1 smoking–related memory with information that will suppress 13 responding to smoking cues. The goal here is to alter existing nicotine-related memory directly rather 14 than rely exclusively on the establishment of an inhibitory extinction process2-6 via traditional cue 15 exposure therapy, which is known to be vulnerable to spontaneous recovery, renewal, and 16 reinstatement7,8. The proposed study will also be innovative and novel because, unlike the Xue et al. 17 2012 study, we will preliminarily assess, over the course of a 1-month follow-up period, the effects of 18 retrieval-extinction training on the smoking behavior of smokers who have made a cessation attempt. 19 20 B. Background and Significance 21 Smoking occurs in approximately 21% of the US population, is responsible for an annual 22 mortality rate of approximately 438,000 citizens, and has an associated healthcare economic burden of 23 $167 billion9. -
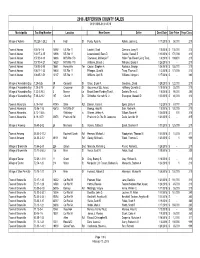
2018 Sales Report
2018 JEFFERSON COUNTY SALES 01/01/2018-01/31/2018 Municipality Tax Map NumberLocation New Owner Seller Deed Date Sale Price Prop Class Village of Adams 112.28-1-28.2 15 High St Purdy, Ryan A. Rohde, Joanne L 1/17/2018$ 58,000 210 Town of Adams 100.14-1-6 14592 US Rte 11 Laisdell, Scott Clemons, Larry R 1/8/2018$ 135,000 210 Town of Adams 100.17-2-15 13906 US Rte 11 Lewandowski, Sean D. Gaulke, Russell S 1/10/2018$ 170,000 210 Town of Adams 107.00-4-13 16686 NYS Rte 178 Townsend, McKenzie F. Haller Fam Revoc Living Trust, 1/8/2018$ 169,600 210 Town of Adams 107.00-4-21 16520 NYS Rte 178 Williams, Brandi J. Williams, Brandi J 1/26/2018$ - 210 Town of Adams 108.05-2-80 18661 Honeyville Ter Casion, Stephen A. Roukous, George 1/26/2018$ 155,000 210 Town of Adams 108.17-1-33 10604 US Rte 11 Pfleegor, Chad M. Tharp, Thomas S 1/3/2018$ 173,000 210 Town of Adams 108.05-1-51 13137 US Rte 11 Williams, April R. Williams, Morgan J 1/17/2018$ - 280 Village of Alexandria Bay 7.29-3-26 39 Cornwall St Olson, Bryan R. Goudreau, David 1/29/2018$ 122,000 210 Village of Alexandria Bay 7.29-3-78 81 Crossmon St Gouverneur S&L Assoc, Hoffberg, Camette C 1/18/2018$ 25,000 210 Village of Alexandria Bay 7.22-2-29.3 3 Mance Ln Broad Street Funding Trust I, Daniels, Emma L 1/5/2018$ 95,000 280 Village of Alexandria Bay 7.38-2-21.2 107 Church St Schreiber, Kenneth M. -

The Effect of Naltrexone and Acamprosate on Cue-Induced
European Neuropsychopharmacology (2007) 17, 558–566 www.elsevier.com/locate/euroneuro The effect of naltrexone and acamprosate on cue-induced craving, autonomic nervous system and neuroendocrine reactions to alcohol-related cues in alcoholics☆ Wendy Ooteman a,b,⁎, Maarten W.J. Koeter a,b, Roel Verheul c,d, Gerard M. Schippers a,b, Wim van den Brink a,b a Amsterdam Institute for Addiction Research, Amsterdam, The Netherlands b Department of Psychiatry, Academic Medical Center University of Amsterdam, Amsterdam, The Netherlands c Viersprong Institute for Studies on Personality Disorders (VISPD), Halsteren, The Netherlands d Department of Clinical Psychology, University of Amsterdam, Amsterdam, The Netherlands Received 22 March 2006; received in revised form 1 February 2007; accepted 13 February 2007 KEYWORDS Abstract Alcoholism; Naltrexone; Introduction: Acamprosate and naltrexone have been shown to be effective in relapse prevention of Acamprosate; alcoholism. It is hypothesized that naltrexone exerts its effects primarily on cue-induced craving Efficacy; and neuroendocrine cue reactivity, whereas acamprosate exerts its effect primarily on autonomic Craving; nervous system reactions to alcohol-related cues. Cue reactivity Experimental procedures: In a randomized double-blind experiment, 131 abstinent alcoholics received either acamprosate (n=56), naltrexone (n=52) or placebo (n=23) for three weeks and participated in two cue-exposure sessions: the first the day before and the second at the last day of medication. Results: Consistent with the hypotheses, naltrexone reduced craving more than acamprosate, and acamprosate reduced heart rate more than naltrexone. No medication effect was found on cue- induced cortisol. Discussion: The findings provide some evidence for differential effects of naltrexone and acamprosate: naltrexone may exert its effect, at least partly, by the reduction of cue-induced craving, whereas acamprosate may exert its effect, at least partly, by the reduction of autonomic nervous system reactions to alcohol-related cues. -

Food Cue Reactivity and Craving Predict Eating and Weight Gain: a Meta-Analytic Review
obesity reviews doi: 10.1111/obr.12354 Etiology and Pathophysiology Food cue reactivity and craving predict eating and weight gain: a meta-analytic review Rebecca G. Boswell1 and Hedy Kober1,2 1Department of Psychology, Yale University, Summary New Haven, CT, USA, and 2Department of According to learning-based models of behavior, food cue reactivity and craving Psychiatry, Yale School of Medicine, Yale are conditioned responses that lead to increased eating and subsequent weight gain. University, New Haven, CT, USA However, evidence supporting this relationship has been mixed. We conducted a quantitative meta-analysis to assess the predictive effects of food cue reactivity Received 16 June 2015; revised 7 October and craving on eating and weight-related outcomes. Across 69 reported statistics 2015; accepted 9 October 2015 from 45 published reports representing 3,292 participants, we found an overall me- dium effect of food cue reactivity and craving on outcomes (r = 0.33, p < 0.001; ap- Address for correspondence: Hedy Kober, proximately 11% of variance), suggesting that cue exposure and the experience of Departments of Psychology and Psychiatry, craving significantly influence and contribute to eating behavior and weight gain. Yale University, 1 Church Street, Suite 701, Follow-up tests revealed a medium effect size for the effect of both tonic and cue- New Haven, CT 06510, USA. induced craving on eating behavior (r = 0.33). We did not find significant differ- E-mail: [email protected] ences in effect sizes based on body mass index, age, or dietary restraint. However, we did find that visual food cues (e.g. -

Iveigh Ivelan Iveland Ivelane Ivelant Ivelen Ivelend Ivelent Ivelind Ivelint
Iveigh Ivones Izearde Izzach Izzox Ivelan Ivoomb Izeart Izzack Izzoyd Iveland Ivoombe Izech Izzacke Izzsard Ivelane Ivor Izeck Izzaech Izzude Ivelant Ivors Izerd Izzaeck Izzume Ivelen Ivory Izert Izzaick Izzyck Ivelend Ivvaren Izett Izzaitch Izzyke Ivelent Ivy Iziac Izzake Ivelind Ivye Izick Izzaock Ivelint Ivymey Izitch Izzaox Ivelyn Ivys Izock Izzard Ivelynd Iwadby Izod Izzarde Ivemey Iwardbey Izode Izzarn Ivemmy Iwardby Izoed Izzarne Ivemy Iwardy Izoit Izzart Iveney Iwerdby Izold Izzarte Ivens Iwoodby Izombe Izzat Iver Ix Izon Izzatt Iverach Ixton Izood Izzayck Iverack Iyon Izoode Izzayke Iveragh Iyone Izoold Izzeard Iverake Iyonne Izoomb Izzearde Iverech Izaac Izoombe Izzeart Ivereck Izaach Izord Izzech Iverick Izaack Izould Izzeck Iveritch Izaacs Izoyd Izzerd Iverock Izaacson Izrael Izzert Iverox Izaake Izraeler Izzett Ivers Izach Izraely Izzick Iverson Izack Izreeli Izzitch Ivery Izacke Izsard Izzock Iveryck Izaech Izsarde Izzod Iveryke Izaeck Izsardt Izzode Ives Izaick Izsart Izzoed Iveson Izaitch Izsarte Izzoit Ivet Izake Izseard Izzold Ivett Izaock Izsearde Izzolm Ivetts Izaox Izseart Izzomb Ivey Izard Izserd Izzombe Ivie Izarde Izsert Izzome Ivimey Izarn Izsord Izzone Ivinges Izarne Izude Izzood Ivington Izart Izyck Izzoode Ivinney Izarte Izyke Izzoold Ivins Izat Izzaac Izzoom Ivinson Izatson Izzaach Izzoomb Iviormy Izatt Izzaack Izzoombe Ivison Izayck Izzaacs Izzord Ivombe Izayke Izzaacson Izzould Ivone Izeard Izzaake Izzown Hall of Names by Swyrich © 1999 Swyrich Corporation www.swyrich.com 1-888-468-7686 557 Jackline Jacok -

Endometrial Response to Conceptus-Derived Estrogen and Interleukin-1Β at the Time of Implantation in Pigs
Ka et al. Journal of Animal Science and Biotechnology (2018) 9:44 https://doi.org/10.1186/s40104-018-0259-8 REVIEW Open Access Endometrial response to conceptus- derived estrogen and interleukin-1β at the time of implantation in pigs Hakhyun Ka1*, Heewon Seo1,2, Yohan Choi1,3, Inkyu Yoo1 and Jisoo Han1 Abstract: The establishment of pregnancy is a complex process that requires a well-coordinated interaction between the implanting conceptus and the maternal uterus. In pigs, the conceptus undergoes dramatic morphological and functional changes at the time of implantation and introduces various factors, including estrogens and cytokines, interleukin-1β2 (IL1B2), interferon-γ (IFNG), and IFN-δ (IFND), into the uterine lumen. In response to ovarian steroid hormones and conceptus-derived factors, the uterine endometrium becomes receptive to the implanting conceptus by changing its expression of cell adhesion molecules, secretory activity, and immune response. Conceptus-derived estrogens act as a signal for maternal recognition of pregnancy by changing the direction of prostaglandin (PG) F2α from the uterine vasculature to the uterine lumen. Estrogens also induce the expression of many endometrial genes, including genes related to growth factors, the synthesis and transport of PGs, and immunity. IL1B2, a pro-inflammatory cytokine, is produced by the elongating conceptus. The direct effect of IL1B2 on endometrial function is not fully understood. IL1B activates the expression of endometrial genes, including the genes involved in IL1B signaling and PG synthesis and transport. In addition, estrogen or IL1B stimulates endometrial expression of IFN signaling molecules, suggesting that estrogen and IL1B act cooperatively in priming the endometrial function of conceptus-produced IFNG and IFND that, in turn, modulate endometrial immune response during early pregnancy. -

Heart Rate Variability As Reactivity in Alcoholics an Index Of
Heart Rate Variability as an Index of Cue Reactivity in Alcoholics I. Rajan, P.J. Naga Venkatesha Murthy, A.G. Ramakrishnan, B.N. Gangadhar, and N. Janakiramaiah Background: Autonomic responses follow exposure to control group on physiological parameters such as skin conditioned stimuli such as contextual factors associated conductance, salivation, heart rate, and peripheral hypo- with alcohol ingestion. Heart rate variabili O, is under auto- thermic response (Drummond et al 1990). nomic control and may be a measure of such response. Spectral analysis of heart rate variability (HRV) is a Methods: Twenty alcoholics and 23 matched social drink- recent noninvasive method of assessing cardiac autonomic ers (all male) were exposed to a neutral cue and then an tone (Malliani et al 1991; Castiglioni 1995). Sympathetic ah'ohol cue in identical settings, during which the elec- and parasympathetic nervous activity is shown to make trocardiogram of these subjects was recorded. Time and frequency-specific contributions to the HRV power spec- frequency domain parameters of heart rate variability trum. Two major components, the high-frequency (HF) (HRV) were computed by a blind rater. and low-frequency (LF) components of the HRV power Results: Coefficient of variation ~( R-R intervals and spectrum, seem to reflect parasympathetic and sympa- absolute powers" of HRV spectrum (in frequency bands thetic activities, respectively (Murata et al 1994). These 0.05-0.15 Hz and 0.01-0.05 Hz) Jbllowing alcohol cue were significantly higher in alcoholics than social drink- sensitive indices of autonomic nervous system filnctioning ers'. The mean heart rate (MHR).fililed to reflect this could be used to measure autonomic perturbations in d~[fl, rence. -

Neurobiology of Craving: Current Findings and New Directions
Current Addiction Reports https://doi.org/10.1007/s40429-018-0202-2 ADOLESCENT/YOUNG ADULT ADDICTION (T CHUNG, SECTION EDITOR) Neurobiology of Craving: Current Findings and New Directions Lara A. Ray1,2 & Daniel J. O. Roche2 # Springer International Publishing AG, part of Springer Nature 2018 Abstract Purpose of the Review This review seeks to provide an update on the current literature on craving and its underlying neurobi- ology, as it pertains to alcohol and drug addiction. Recent Findings Studies on craving neurobiology suggest that the brain networks activated by conditioned cues in alcohol- and drug-dependent populations extend far beyond the traditional mesolimbic dopamine system and suggest that the early neurobiolog- ical theories of addiction, which heavily relied on dopamine release into the nucleus accumbens as the primary mechanism driving cue-induced craving and drug-seeking behavior, are incomplete. Ongoing studies will advance our understanding of the neurobio- logical underpinnings of addiction and drug craving by identifying novel brain regions associated with responses to conditioned cues that may be specific to humans, or at least primates, due to these brain areas’ involvement in higher cognitive processes. Summary This review highlights recent advances and future directions in leveraging the neurobiology of craving as a transla- tional phenotype for understanding addiction etiology and informing treatment development. The complexity of craving and its underlying neurocircuitry is evident and divergent methods of eliciting craving (i.e., cues, stress, and alcohol administration) may produce divergent findings. Keywords Craving . Addiction . Neurobiology . fMRI . Cue reactivity . Alcohol . Drug Introduction relative risk of all other diagnostic criteria for alcoholism [4]. -

Okhrana Records
http://oac.cdlib.org/findaid/ark:/13030/kt538nf189 Online items available Register of the Okhrana records Finding aid prepared by Andrej Kobal and Sally DeBauche Hoover Institution Archives 434 Galvez Mall Stanford University Stanford, CA, 94305-6010 (650) 723-3563 [email protected] © 1964, 2016 Register of the Okhrana records 26001 1 Title: Okhrana records Date (inclusive): 1883-1917 Collection Number: 26001 Contributing Institution: Hoover Institution Archives Language of Material: Russian Physical Description: 232 manuscript boxes, 86 card file boxes, 6 oversize boxes(194.6 linear feet) Abstract: Intelligence reports from agents in the field and the Paris office, dispatches, circulars, headquarters studies, correspondence of revolutionaries, and photographs, relating to activities of Russian revolutionists abroad. Collection is available on microfilm (509 reels). Digital copies of select records also available at https://digitalcollections.hoover.org. Physical Location: Hoover Institution Archives Creator: Russia. Departament politsii. Zagranichnaia agentura (Paris) Access Microfilm use only. Digital copies of select records also available at https://digitalcollections.hoover.org. The Hoover Institution Archives only allows access to copies of audiovisual items. To listen to sound recordings or to view videos or films during your visit, please contact the Archives at least two working days before your arrival. We will then advise you of the accessibility of the material you wish to see or hear. Please note that not all audiovisual material is immediately accessible. Publication Rights For copyright status, please contact the Hoover Institution Archives. Preferred Citation [Identification of item], Okhrana records, [Index number, Folder number], Hoover Institution Archives. Acquisition Information Acquired by the Hoover Institution Archives in 1926. -

Urine-Derived Epithelial Cells As Models for Genetic Kidney Diseases
cells Review Urine-Derived Epithelial Cells as Models for Genetic Kidney Diseases Tjessa Bondue 1 , Fanny O. Arcolino 1 , Koenraad R. P. Veys 1,2, Oyindamola C. Adebayo 1,3, Elena Levtchenko 1,2, Lambertus P. van den Heuvel 1,4 and Mohamed A. Elmonem 5,* 1 Department of Development and Regeneration, KU Leuven, 3000 Leuven, Belgium; [email protected] (T.B.); [email protected] (F.O.A.); [email protected] (K.R.P.V.); [email protected] (O.C.A.); [email protected] (E.L.); [email protected] (L.P.v.d.H.) 2 Department of Pediatrics, Division of Pediatric Nephrology, University Hospitals Leuven, 3000 Leuven, Belgium 3 Centre for Molecular and Vascular Biology, Department of Cardiovascular Sciences, KU Leuven, 3000 Leuven, Belgium 4 Department of Pediatric Nephrology, Radboud University Medical Center, 6500 Nijmegen, The Netherlands 5 Department of Clinical and Chemical Pathology, Faculty of Medicine, Cairo University, Cairo 11628, Egypt * Correspondence: [email protected] Abstract: Epithelial cells exfoliated in human urine can include cells anywhere from the urinary tract and kidneys; however, podocytes and proximal tubular epithelial cells (PTECs) are by far the most relevant cell types for the study of genetic kidney diseases. When maintained in vitro, they have been proven extremely valuable for discovering disease mechanisms and for the development of new therapies. Furthermore, cultured patient cells can individually represent their human sources and their specific variants for personalized medicine studies, which are recently gaining much Citation: Bondue, T.; Arcolino, F.O.; interest. In this review, we summarize the methodology for establishing human podocyte and PTEC Veys, K.R.P.; Adebayo, O.C.; cell lines from urine and highlight their importance as kidney disease cell models.