Cycads, Flying Foxes, and Brain Disease in Humans
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
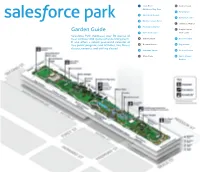
Salesforce Park Garden Guide
Start Here! D Central Lawn Children’s Play Area Garden Guide6 Palm Garden 1 Australian Garden Start Here! D Central Lawn Salesforce Park showcases7 California over Garden 50 species of Children’s Play Area 2 Mediterraneantrees and Basin over 230 species of understory plants. 6 Palm Garden -ã ¼ÜÊ ÊăØÜ ØÊèÜãE úØƀØÊèÃJapanese Maples ¼ÃØ Ê¢ 1 Australian Garden 3 Prehistoric¢ØÕ輫ÕØÊ£ØÂÜÃã«ó«ã«Üŧ¼«¹ĆãÃÜÜ Garden 7 California Garden ¼ÜÜÜŧÊÃØãÜŧÃØ¢ã«Ã£¼ÜÜÜũF Amphitheater Garden Guide 2 Mediterranean Basin 4 Wetland Garden Main Lawn E Japanese Maples Salesforce Park showcases over 50 species of 3 Prehistoric Garden trees and over 230 species of understory plants. A Oak Meadow 8 Desert Garden F Amphitheater It also offers a robust year-round calendar of 4 Wetland Garden Main Lawn free public programs and activities, like fitness B Bamboo Grove 9 Fog Garden Desert Garden classes, concerts, and crafting classes! A Oak Meadow 8 5 Redwood Forest 10 Chilean Garden B Bamboo Grove 9 Fog Garden C Main Plaza 11 South African 10 Chilean Garden Garden 5 Redwood Forest C Main Plaza 11 South African Garden 1 Children’s Australian Play Area Garden ABOUT THE GARDENS The botanist aboard the Endeavor, Sir Joseph Banks, is credited with introducing many plants from Australia to the western world, and many This 5.4 acre park has a layered soil system that plants today bear his name. balances seismic shifting, collects and filters storm- water, and irrigates the gardens. Additionally, the soil Native to eastern Australia, Grass Trees may grow build-up and dense planting help offset the urban only 3 feet in 100 years, and mature plants can be heat island effect by lowering the air temperature. -

Chemical Hygiene Plan Manual
CHEMICAL HYGIENE PLAN AND HAZARDOUS MATERIALS SAFETY MANUAL FOR LABORATORIES This is the Chemical Hygiene Plan specific to the following areas: Laboratory name or room number(s): ___________________________________ Building: __________________________________________________________ Supervisor: _______________________________________________________ Department: _______________________________________________________ Telephone numbers 911 for Emergency and urgent consultation 48221 Police business line 46919 Fire Dept business line 46371 Radiological and Environmental Management Revisied on: Enter a revision date here. All laboratory chemical use areas must maintain a work-area specific Chemical Hygiene Plan which conforms to the requirements of the OSHA Laboraotry Standard 29 CFR 19190.1450. Purdue University laboratories may use this document as a starting point for creating their work area specific CHP. Minimally this cover page is to be edited for work area specificity (non-West Lafayette laboratories are to place their own emergency, fire, and police telephone numbers in the space above) AND appendix K must be completed. This instruction and information box should remain. This model CHP is version 2010A; updates are to be found at www.purdue.edu/rem This page intentionally blank. PURDUE CHEMICAL HYGIENE PLAN AWARENESS CERTIFICATION For CHP of: ______________________________ Professor, building, rooms The Occupational Safety and Health Administration (OSHA) requires that laboratory employees be made aware of the Chemical Hygiene Plan at their place of employment (29 CFR 1910.1450). The Purdue University Chemical Hygiene Plan and Hazardous Materials Safety Manual serves as the written Chemical Hygiene Plan (CHP) for laboratories using chemicals at Purdue University. The CHP is a regular, continuing effort, not a standby or short term activity. Departments, divisions, sections, or other work units engaged in laboratory work whose hazards are not sufficiently covered in this written manual must customize it by adding their own sections as appropriate (e.g. -

Bush Foods and Fibres
Australian Plants Society NORTH SHORE GROUP Ku-ring-gai Wildflower Garden Bush foods and fibres • Plant-based bush foods, medicines and poisons can come from nectar, flowers, fruit, leaves, bark, stems, sap and roots. • Plants provide fibres and materials for making many items including clothes, cords, musical instruments, shelters, tools, toys and weapons. • A fruit is the seed-bearing structure of a plant. • Do not eat fruits that you do not know to be safe to eat. Allergic reactions or other adverse reactions could occur. • We acknowledge the Traditional Custodians of this land and pay our respects to the Elders both past, present and future for they hold the memories, traditions, culture and hope of their people. Plants as food: many native plants must be processed before they are safe to eat. Flowers, nectar, pollen, Sugars, vitamins, honey, lerps (psyllid tents) minerals, starches, manna (e.g. Ribbon Gum proteins & other nutrients Eucalyptus viminalis exudate), gum (e.g. Acacia lerp manna decurrens) Fruit & seeds Staple foods Carbohydrates (sugars, starches, fibre), proteins, fats, vitamins Leaves, stalks, roots, apical Staple foods Carbohydrates, protein, buds minerals Plants such as daisies, lilies, orchids and vines Tubers, rhyzomes were a source of starchy tubers known as Carbohydrate, fibre, yams. The yam daisy Microseris lanceolata protein, vitamins, (Asteraceae) was widespread in inland NSW minerals and other states. The native yam Dioscorea transversa grows north from Stanwell Tops into Qld and Northern Territory and can be eaten raw or roasted as can those of Trachymene incisa. 1 Plant Description of food Other notes Acacia Wattle seed is a rich source of iron, Saponins and tannins and other essential elements. -

Complexity and Variation in the Effects of Low-Severity Fires on Forest Biota
University of Montana ScholarWorks at University of Montana Graduate Student Theses, Dissertations, & Professional Papers Graduate School 2003 Complexity and variation in the effects of low-severity fires on forest biota Karen Christine Short The University of Montana Follow this and additional works at: https://scholarworks.umt.edu/etd Let us know how access to this document benefits ou.y Recommended Citation Short, Karen Christine, "Complexity and variation in the effects of low-severity fires on forest biota" (2003). Graduate Student Theses, Dissertations, & Professional Papers. 9463. https://scholarworks.umt.edu/etd/9463 This Dissertation is brought to you for free and open access by the Graduate School at ScholarWorks at University of Montana. It has been accepted for inclusion in Graduate Student Theses, Dissertations, & Professional Papers by an authorized administrator of ScholarWorks at University of Montana. For more information, please contact [email protected]. Maureen and Mike MANSFIELD LIBRARY The University of Montana Permission is granted by the author to reproduce this material in its entirety, provided that this material is used for scholarly purposes and is properly cited in published works and reports. **Please check "Yes" or "No" and provide signature** ,/ Yes, I grant permission U7 ______ No, I do not grant permission Author's Signature: Date: <£/£-/< Any copying for commercial purposes or financial gain may be undertaken only with the author's explicit consent. 8/98 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. COMPLEXITY AND VARIATION IN THE EFFECTS OF LOW-SEVERITY FIRES ON FOREST BIOTA by Karen C. -

Brisbane Native Plants by Suburb
INDEX - BRISBANE SUBURBS SPECIES LIST Acacia Ridge. ...........15 Chelmer ...................14 Hamilton. .................10 Mayne. .................25 Pullenvale............... 22 Toowong ....................46 Albion .......................25 Chermside West .11 Hawthorne................. 7 McDowall. ..............6 Torwood .....................47 Alderley ....................45 Clayfield ..................14 Heathwood.... 34. Meeandah.............. 2 Queensport ............32 Trinder Park ...............32 Algester.................... 15 Coopers Plains........32 Hemmant. .................32 Merthyr .................7 Annerley ...................32 Coorparoo ................3 Hendra. .................10 Middle Park .........19 Rainworth. ..............47 Underwood. ................41 Anstead ....................17 Corinda. ..................14 Herston ....................5 Milton ...................46 Ransome. ................32 Upper Brookfield .......23 Archerfield ...............32 Highgate Hill. ........43 Mitchelton ...........45 Red Hill.................... 43 Upper Mt gravatt. .......15 Ascot. .......................36 Darra .......................33 Hill End ..................45 Moggill. .................20 Richlands ................34 Ashgrove. ................26 Deagon ....................2 Holland Park........... 3 Moorooka. ............32 River Hills................ 19 Virginia ........................31 Aspley ......................31 Doboy ......................2 Morningside. .........3 Robertson ................42 Auchenflower -

Rdna) Organisation
OPEN Heredity (2013) 111, 23–33 & 2013 Macmillan Publishers Limited All rights reserved 0018-067X/13 www.nature.com/hdy ORIGINAL ARTICLE Dancing together and separate again: gymnosperms exhibit frequent changes of fundamental 5S and 35S rRNA gene (rDNA) organisation S Garcia1 and A Kovarˇı´k2 In higher eukaryotes, the 5S rRNA genes occur in tandem units and are arranged either separately (S-type arrangement) or linked to other repeated genes, in most cases to rDNA locus encoding 18S–5.8S–26S genes (L-type arrangement). Here we used Southern blot hybridisation, PCR and sequencing approaches to analyse genomic organisation of rRNA genes in all large gymnosperm groups, including Coniferales, Ginkgoales, Gnetales and Cycadales. The data are provided for 27 species (21 genera). The 5S units linked to the 35S rDNA units occur in some but not all Gnetales, Coniferales and in Ginkgo (B30% of the species analysed), while the remaining exhibit separate organisation. The linked 5S rRNA genes may occur as single-copy insertions or as short tandems embedded in the 26S–18S rDNA intergenic spacer (IGS). The 5S transcript may be encoded by the same (Ginkgo, Ephedra) or opposite (Podocarpus) DNA strand as the 18S–5.8S–26S genes. In addition, pseudogenised 5S copies were also found in some IGS types. Both L- and S-type units have been largely homogenised across the genomes. Phylogenetic relationships based on the comparison of 5S coding sequences suggest that the 5S genes independently inserted IGS at least three times in the course of gymnosperm evolution. Frequent transpositions and rearrangements of basic units indicate relatively relaxed selection pressures imposed on genomic organisation of 5S genes in plants. -

Garden Plants Poisonous to People
N NO V E M B E R 2 0 0 6 P R I M E F A C T 3 5 9 ( R E P L A C E S A G F A C T P 7 . 1 . 1 P O I S O N O U S P L A N T S I N T H E G A R D E N) Garden plants poisonous to people Annie Johnson Table 1. Toxicity rating for Tables 2−7. Weeds Project Officer Rating Toxicity Stephen Johnson Mildly toxic. Mild symptoms may occur if large * Weed Ecologist quantities are eaten. Toxic. Causes discomfort and irritation but not Weeds Unit, Biosecurity Compliance and Mine ** Safety, Orange dangerous to life. Highly toxic. Capable of causing serious illness *** or death. Introduction There are a range of garden plants that are considered poisonous. Poisonings and deaths from garden plants Poisoning are rare as most poisonous plants taste unpleasant Poisoning from plants may occur from ingesting, and are seldom swallowed (see toxicity). However, it is inhalation or direct contact. best to know which plants are potentially toxic. Symptoms from ingestion include gastroenteritis, It is important to remember that small children are diarrhoea, vomiting, nervous symptoms and in serious often at risk from coloured berries, petals and leaves cases, respiratory and cardiac distress. Poisoning that look succulent. This does not mean that all these by inhalation of pollen, dust or fumes from burning poisonous plants should be avoided or removed from plants can cause symptoms similar to hay fever or the garden. It is best to teach children never to eat asthma. -

Ghs Reference Materials Health Hazard Criteria
APPENDIX F: GHS REFERENCE MATERIALS This appendix provides both an overview of GHS highly toxic hazard classification. A listing of Particularly Hazardous Substances that Carnegie Mellon University has published with their Chemical Hygiene plan is also provided as general reference. The list is useful cross check with GHS listings to determine which materials require prior approval for use BUT NO LIST IS COMPLETE you must check the SDS for possible additional chemicals rated as highly toxic. This appendix also provides the GHS (global harmonization system) for chemical hazard classification under the Hazard Communication Standard for highly toxic materials. This section provides overall information about categories under the classification of acute toxicity, mutagens’, reproductive and carcinogen hazards. When chemicals are rated on the GHS – Safety Data Sheet (SDS) as the following hazards then the PRIOR APPROVAL PROCESS WITH CHEMICAL HYGIENE OFFICER/COMMITTEE must be used: Acute toxicity category 1 and 2, Germ cell mutagenicity as a category 1A Substances known to induce heritable mutations in germ cells of humans and Category 1B: Substances which should be regarded as if they induce heritable mutations in the germ cells of humans, Reproductive Hazard as a category 1: Known or presumed human reproductive toxicants and Category 2; suspected human reproductive toxicant. Carcinogen as a Category 1 (includes 1A and 1B): Known or presumed human carcinogens, Category 2: Suspected human carcinogens. The Campus Chemical Hygiene Committee (Officer) must conduct a prior approval process. Appendix C Chemical Prior Approval Form on procedure for conducing prior approval. The following is from OSHA standard on the chemicals classifications that PCC Laboratory instructional operations shall use for defining the prior approval hazards. -

Elemental Profiles in Cycas Micronesica Stems
plants Article Elemental Profiles in Cycas micronesica Stems Thomas E. Marler College of Natural and Applied Sciences, University of Guam, UOG Station, Mangilao, Guam 96923, USA; [email protected]; Tel.: +1-671-735-2100 Received: 24 August 2018; Accepted: 30 October 2018; Published: 1 November 2018 Abstract: Essential nutrients and metals have been quantified in stems of many tree species to understand the role of stems as storage and source organs. Little is known about stored stem resources of cycad tree species. Cycas micronesica tissue was collected from apical and basal axial regions of stems; and pith, vascular, and cortex tissues were separated into three radial regions. Leaves were also sampled to provide a comparison to stems. Minerals and metals were quantified in all tissues. Minerals and metals varied greatly among the six stem sections. Phosphorus varied more among the three radial sections than the other macronutrients, and zinc and nickel varied more than the other micronutrients. Stem carbon was less than and stem calcium was greater than expected, based on what is currently known tree stem concentrations in the literature. Elemental concentrations were generally greater than those previously reported for coniferous gymnosperm trees. Moreover, the stem concentrations were high in relation to leaf concentrations, when compared to published angiosperm and conifer data. The results indicated that the addition of more cycad species to the literature will improve our understanding of gymnosperm versus angiosperm stem nutrient relations, and that the non-woody cycad stem contains copious essential plant nutrients that can be mobilized and deployed to sinks when needed. -

Vegetation and Flora of Booti Booti National Park and Yahoo Nature Reserve, Lower North Coast of New South Wales
645 Vegetation and flora of Booti Booti National Park and Yahoo Nature Reserve, lower North Coast of New South Wales. S.J. Griffith, R. Wilson and K. Maryott-Brown Griffith, S.J.1, Wilson, R.2 and Maryott-Brown, K.3 (1Division of Botany, School of Rural Science and Natural Resources, University of New England, Armidale NSW 2351; 216 Bourne Gardens, Bourne Street, Cook ACT 2614; 3Paynes Lane, Upper Lansdowne NSW 2430) 2000. Vegetation and flora of Booti Booti National Park and Yahoo Nature Reserve, lower North Coast of New South Wales. Cunninghamia 6(3): 645–715. The vegetation of Booti Booti National Park and Yahoo Nature Reserve on the lower North Coast of New South Wales has been classified and mapped from aerial photography at a scale of 1: 25 000. The plant communities so identified are described in terms of their composition and distribution within Booti Booti NP and Yahoo NR. The plant communities are also discussed in terms of their distribution elsewhere in south-eastern Australia, with particular emphasis given to the NSW North Coast where compatible vegetation mapping has been undertaken in many additional areas. Floristic relationships are also examined by numerical analysis of full-floristics and foliage cover data for 48 sites. A comprehensive list of vascular plant taxa is presented, and significant taxa are discussed. Management issues relating to the vegetation of the reserves are outlined. Introduction The study area Booti Booti National Park (1586 ha) and Yahoo Nature Reserve (48 ha) are situated on the lower North Coast of New South Wales (32°15'S 152°32'E), immediately south of Forster in the Great Lakes local government area (Fig. -

Detection of Cyanotoxins, Β-N-Methylamino-L-Alanine and Microcystins, from a Lake Surrounded by Cases of Amyotrophic Lateral Sclerosis
Toxins 2015, 7, 322-336; doi:10.3390/toxins7020322 OPEN ACCESS toxins ISSN 2072-6651 www.mdpi.com/journal/toxins Article Detection of Cyanotoxins, β-N-methylamino-L-alanine and Microcystins, from a Lake Surrounded by Cases of Amyotrophic Lateral Sclerosis Sandra Anne Banack 1, Tracie Caller 2,†, Patricia Henegan 3,†, James Haney 4,†, Amanda Murby 4, James S. Metcalf 1, James Powell 1, Paul Alan Cox 1 and Elijah Stommel 3,* 1 Institute for Ethnomedicine, PO Box 3464, Jackson, WY 83001, USA; E-Mails: [email protected] (S.A.B.); [email protected] (J.S.M.); [email protected] (J.P.); [email protected] (P.A.C.) 2 Cheyenne Regional Medical Group, Cheyenne, WY 82001, USA; E-Mail: [email protected] 3 Department of Neurology, Dartmouth-Hitchcock Medical Center, Lebanon, NH 03756, USA; E-Mail: [email protected] 4 Department of Biological Sciences, University of New Hampshire, Durham, NH 03824, USA; E-Mails: [email protected] (J.H.); [email protected] (A.M.) † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-603-650-8615; Fax: +1-603-650-6233. Academic Editor: Luis M. Botana Received: 19 November 2014 / Accepted: 21 January 2015 / Published: 29 January 2015 Abstract: A cluster of amyotrophic lateral sclerosis (ALS) has been previously described to border Lake Mascoma in Enfield, NH, with an incidence of ALS approximating 25 times expected. We hypothesize a possible association with cyanobacterial blooms that can produce β-N-methylamino-L-alanine (BMAA), a neurotoxic amino acid implicated as a possible cause of ALS/PDC in Guam. -

National Multi-Species Recovery Plan for the Cycads, Cycas Megacarpa
National Multi-species Recovery Plan for the cycads, Cycas megacarpa, Cycas ophiolitica, Macrozamia cranei, Macrozamia lomandroides, Macrozamia pauli-guilielmi and Macrozamia platyrhachis Prepared by the Queensland Herbarium, Environmental Protection Agency, Brisbane Cycas megacarpa male (photo: P.I. Forster) Cycas ophiolitica female (photo: G.W. Wilson) Macrozamia cranei male (photo: P.I. Forster) Macrozamia lomandroides female (photo: G. Leiper) Macrozamia pauli-guilielmi male and female growing together (photo: G. Leiper) Macrozamia platyrhachis male (photo: P.I. Forster) National Multi-species Recovery Plan for the cycads, Cycas megacarpa, Cycas ophiolitica, Macrozamia cranei, Macrozamia lomandroides, Macrozamia pauli-guilielmi and Macrozamia platyrhachis Prepared by: Paul Forster and Ailsa Holland of the Queensland Herbarium, Environmental Protection Agency. © The State of Queensland, Environmental Protection Agency Copyright protects this publication. Except for purposes permitted by the Copyright Act, reproduction by whatever means is prohibited without the prior written knowledge of the Environmental Protection Agency. Inquiries should be addressed to PO Box 15155, CITY EAST, QLD 4002. Copies may be obtained from the: Executive Director Conservation Services Queensland Parks and Wildlife Service PO Box 15155 City East QLD 4002 Disclaimer: The Australian Government, in partnership with the Environmental Protection Agency/Queensland Parks and Wildlife Service, facilitates the publication of recovery plans to detail the actions needed for the conservation of threatened native wildlife. The attainment of objectives and the provision of funds may be subject to budgetary and other constraints affecting the parties involved, and may also be constrained by the need to address other conservation priorities. Approved recovery actions may be subject to modification due to changes in knowledge and changes in conservation status.