Electroanalytical Methods for Determination of Sunset Yellow— a Review
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Artificial Food Colours and Children Why We Want to Limit and Label Foods Containing the ‘Southampton Six’ Food Colours on the UK Market Post-Brexit
Artificial food colours and children Why we want to limit and label foods containing the ‘Southampton Six’ food colours on the UK market post-Brexit November 2020 FIRST STEPS NUTRITIONArtificial food coloursTRUST and children: page Artificial food colours and children: Why we want to limit and label foods containing the‘Southampton Six’ food colours on the UK market post-Brexit November 2020 Published by First Steps Nutrition Trust. A PDF of this resource is available on the First Steps Nutrition Trust website. www.firststepsnutrition.org The text of this resource, can be reproduced in other materials provided that the materials promote public health and make no profit, and an acknowledgement is made to First Steps Nutrition Trust. This resource is provided for information only and individual advice on diet and health should always be sought from appropriate health professionals. First Steps Nutrition Trust Studio 3.04 The Food Exchange New Covent Garden Market London SW8 5EL Registered charity number: 1146408 First Steps Nutrition Trust is a charity which provides evidence-based and independent information and support for good nutrition from pre-conception to five years of age. For more information, see our website: www.firststepsnutrition.org Acknowledgements This report was written by Rachael Wall and Dr Helen Crawley. We would like to thank Annie Seeley, Sarah Weston, Erik Millstone and Anna Rosier for their help and support with this report. Artificial food colours and children: page 1 Contents Page Executive summary 3 Recommendations -

Global Regulations of Food Colors Each Region Has Its Own Definitions of What Constitutes a Color Additive, with Related Use Requirements and Restrictions
Global Regulations of Food Colors Each region has its own definitions of what constitutes a color additive, with related use requirements and restrictions. Sue Ann McAvoy Sensient Colors LLC t is said, we eat with our eyes . Since antiq - food colors was soon recognized as a threat Iuity, humans have used the color of a to public health. Of concern was that some food to discern its quality. Color provides of the substances were known to be poi - a way to judge ripeness, perceive flavor and sonous and were often incorporated to hide assess quality of food. poor quality, add bulk to foods and to pass Ancient civilizations introduced color off imitation foods as real. into their foods. The ancient Egyptians col - On February 1, 1899, the executive com - ored their food yellow with saffron, and the mittee of the National Confectioners’ Asso - ancient Mayans used annatto to color their ciation published an official circular which Sue Ann McAvoy is food orange-red. Wealthy Romans ate bread was “to throw light upon the vexed question global regulatory scien - that had been whitened by adding alum to of what colors may be safely used in confec - tist for Sensient Food the flour. Color could be used to enhance tionery” as “there may at times be a doubt in Colors LLC. She has worked at Sensient the mind of the honest confectioner as to the physical appearance of the product. since 1979. However, if it made the food appear to be which colors, flavors, or ingredients he may of better quality than it was, that was con - safely use and which he may reject.” This list sidered a deceitful practice. -

Regulatory Information Sheet
Regulatory Information Sheet Approved Drug Colourants Listed by the European Union Colour Index Colour E Number Alternate Names Number Allura Red AC (a) E129 16035 FD&C Red #40 Aluminum*** E173 77000 -- Amaranth*** (a) E123 16185 Delisted FD&C Red #2 Annatto*** E160b 75120 Bixin, norbixin Anthocyanins (a) E163 -- -- Beetroot Red E162 -- Betanin Beta APO-8´-Carotenal E160e 40820 -- Brilliant Black BN (a) E151 28440 Black BN Brilliant Blue FCF (a) E133 42090 FD&C Blue #1 Brown HT (a) E155 20285 -- Calcium Carbonate E170 77220 -- Canthaxanthin* E161g 40850 -- Caramel,-Plain E150a -- -- Caramel,-Caustic Sulphite E150b -- -- Caramel,-Ammonia E150c -- -- Caramel, Sulphite Ammonia E150d -- -- Carmine (a) E120 75470 Carminic Acid, Cochineal Carmoisine (a) E122 14720 Azorubine Carotenes E160a 40800 / 75130 -- Chlorophylls/Chlorophyllins E140 75810 / 75815 -- Copper Complexes of E141 75815 -- Chlorophylls/Chlorophyllins(a) Curcumin (a) E100 75300 Turmeric Erythrosine*** (a) E127 45430 FD&C Red #3 Gold*** E175 77480 -- Green S (a) E142 44090 Acid Brilliant Green BS Indigotine (a) E132 73015 FD&C Blue #2, Indigo Carmine 77491 / 77492 / Iron Oxides & Hydroxides E172 Iron Oxide Red, Yellow, Black 77499 Litholrubine BK*** (a) E180 -- -- Lutein E161b -- -- Lycopene*** E160d 75125 -- Paprika Extract E160c -- Capsanthin, Capsorubin Patent Blue V (a) E131 42051 Acid Blue 3 Ponceau 4R (a) E124 16255 Cochineal Red A Page 1 of 2 Document Reference No.: GLO-10107, revision 2 Effective Date: September 2014 Reviewed Date: November 2017 This document is valid at the time of distribution. Distributed 24-Sep-2021 (UTC) E Colour Index Colour Alternate Names Number Number Quinoline Yellow** (a) E104 47005 China Yellow Riboflavins (a) E101 -- -- Silver*** E174 -- -- Sunset Yellow FCF (a) E110 15985 FD&C Yellow #6, Orange Yellow S Tartrazine (a) E102 19140 FD&C Yellow #5 Titanium Dioxide E171 77891 -- Vegetable Carbon E153 77268:1 Carbo Medicinalis Vegetalis The above list is derived from Part B, List of All Additives, from Annex II to Regulation (EC) No 1333/2008 on food additives. -

Spectrophotometric Determination of Sunset Yellow (E-110) in Powdered Beverages and Pharmaceutical Preparations After Cloud Point Extraction Method
Güray T. JOTCSA. 2018; 5(2): 479-492. RESEARCH ARTICLE Spectrophotometric Determination of Sunset Yellow (E-110) in Powdered Beverages and Pharmaceutical Preparations after Cloud Point Extraction Method Tufan GÜRAY1* 1Eskisehir Osmangazi University, Faculty of Arts and Science, Department of Chemistry, 26480, Eskisehir, Turkey. Abstract: In this study, Brij 58 was used for the spectrophotometric determination of sunset yellow (SY) (E-110) in pharmaceutical preparations and powdered beverages after cloud point extraction (CPE). Certain parameters such as pH, surfactant concentration, extraction time and temperature, speed of centrifugation, and salt concentration were optimized. Linear range in the optimum conditions was 0.01 – 4.00g mL-1 and the correlation coefficient was 0.9995. The limit of detection (LOD) and the limit of quantification (LOQ) of this method were 0.0078g mL- 1 and 0.0261 g mL-1, respectively. Keywords: Brij 58; Sunset Yellow (E-110); Cloud Point Extraction (CPE); Spectrophotometric Determination; Surfactant Submitted: November 05, 2017. Accepted: March 04, 2018. Cite this: Güray T. Spectrophotometric Determination of Sunset Yellow (E-110) in Powdered Beverages and Pharmaceutical Preparations after Cloud Point Extraction Method. JOTCSA. 2018;5(2):479–92. DOI: http://dx.doi.org/10.18596/jotcsa.349382. *Corresponding author. E-mail: [email protected]; [email protected] 479 Güray T. JOTCSA. 2018; 5(2): 479-492. RESEARCH ARTICLE INTRODUCTION Food dyes are used to provide more attractive, appetizing appearances to enhance the taste, flavor, and color of foods (1). Food coloring Sunset Yellow (6-hydroxy-5- [(4-sulfophenyl) azo] -2-naphthalenesulfonic acid disodium salt) (SY) (E-110) is a food additive commonly used in foods, pharmaceuticals, and cosmetics. -

Organic Colouring Agents in the Pharmaceutical Industry
DOI: 10.1515/fv-2017-0025 FOLIA VETERINARIA, 61, 3: 32—46, 2017 ORGANIC COLOURING AGENTS IN THE PHARMACEUTICAL INDUSTRY Šuleková, M.1, Smrčová, M.1, Hudák, A.1 Heželová, M.2, Fedorová, M.3 1Department of Chemistry, Biochemistry and Biophysics, Institute of Pharmaceutical Chemistry University of Veterinary Medicine and Pharmacy, Komenského 73, 041 81 Košice 2Faculty of Metallurgy, Institute of Recycling Technologies Technical University in Košice, Letná 9, 042 00 Košice 3Department of Pharmacy and Social Pharmacy University of Veterinary Medicine and Pharmacy, Komenského 73, 041 81 Košice Slovakia [email protected] ABSTRACT INTRODUCTION Food dyes are largely used in the process of manufac- In addition to the active ingredients, various additives turing pharmaceutical products. The aim of such a pro- are used in the manufacture of pharmaceuticals. This group cedure is not only to increase the attractiveness of prod- of compounds includes dyes. A colour additive is any dye, ucts, but also to help patients distinguish between phar- pigment, or other substance that imparts colour to food, maceuticals. Various dyes, especially organic colouring drink or any non-food applications including pharma- agents, may in some cases have a negative impact on the ceuticals. Moreover, a colour additive is also any chemical human body. They are incorporated into pharmaceuti- compound that reacts with another substance and causes cal products including tablets, hard gelatine capsules or the formation of a colour [22, 56]. The pharmaceutical in- soft gelatine capsules, lozenges, syrups, etc. This article dustry employs various inorganic and, especially, organic provides an overview of the most widely used colouring dyes for this purpose. -

Analysis of Artificial Colorants in Various Food Samples Using Monolithic Silica Columns and LC-MS by Stephan Altmaier, Merck Millipore, Frankfurter Str
31 Analysis of Artificial Colorants in Various Food Samples using Monolithic Silica Columns and LC-MS by Stephan Altmaier, Merck Millipore, Frankfurter Str. 250, 64293 Darmstadt, Germany This work describes a simple and sensitive high performance liquid chromatography method with UV or mass spectrometry detection for the analysis of artificial colorants from dye classes such as azo or chinophthalone in various food samples. After a short sample preparation procedure all samples were separated on C18 reversed phase monolithic silica columns via a gradient elution profile and directly transferred to UV or MS for the analysis of the main components. This setup enabled the identification of dyes in real life samples such as beverages or sweets within very short analysis times and with a minimised sample preparation step. In the nineteenth century chemicals such as azo compounds (see Tables 1 and 2 and by an organism very easily. mercury sulphide, lead oxide, copper salts or Figure 1). They are utilised as a single Most of the current artificial colorants can now fuchsine were utilised to artificially colour colouring ingredient or as a mixture with be replaced by natural dyes very easily. food such as cheese, confectionary, pickles other colorants in a wide variety of foods and Nevertheless, for economic reasons they are [1] or wine [2]. In the end of that century the beverages. All listed dyes are nontoxic and still used to improve the attractiveness of discovery of many synthetic organic food water soluble and can therefore be excreted sweets or soft drinks towards children or of colorants allowed for more brilliant colours than traditional natural dyes. -

Domestic and Import Food Additives and Color Additives
FOOD AND DRUG ADMINISTRATION COMPLIANCE PROGRAM GUIDANCE MANUAL PROGRAM 7309.006 CHAPTER 09 – FOOD AND COLOR ADDITIVES SUBJECT: IMPLEMENTATION DATE: DOMESTIC AND IMPORT FOOD ADDITIVES AND 10/28/2019 COLOR ADDITIVES DATA REPORTING PRODUCT CODES PRODUCT/ASSIGNMENT CODES All Food Codes (except Industry 16 09006C Color Additives (seafood)) and Industry 45-46 (Food Additives) 09006F Food Additives All Food Codes (except Industry 16 (seafood)) and Industry 50 (Color Additives) FIELD REPORTING REQUIREMENTS: Report all sample collections and analytical results into the Field Accomplishment and Compliance Tracking System (FACTS). Report all inspections into eNSpect. Scan product labeling and any product brochures into eNSpect as an exhibit. Date of Issuance: 10/28/2019 Page 1 of 2 PROGRAM 7309.006 Contents PART I - BACKGROUND .................................................................................................................... 3 PART II - IMPLEMENTATION............................................................................................................ 6 Objectives .................................................................................................................................... 6 Program Management Instructions .............................................................................................. 6 PART III - INSPECTIONAL ................................................................................................................. 9 Operations ................................................................................................................................... -

FCC/USP Survey on Synthetic Food Color Additives
FCC/USP Survey on Synthetic Food Color Additives Background and Purpose: The Food Chemicals Codex (FCC) is a compendium of internationally recognized testing standards owned by US Pharmacopeia for assessing the authenticity, quality, purity, and thus safety of food ingredients. In an effort to update and modernize FCC testing standards for synthetic food color additives, FCC is seeking up-to-date information from leading producers of these ingredients on the chemical composition and analytical test methods for the eighteen color additives listed below in Table 1, along with samples of individual color additives or impurities. Information collected by FCC will be used to update existing monograph testing standards for these color additives or for the creation of new testing standards for those not already in FCC. Samples collected by FCC will be used by USP labs to evaluate candidate analytical methods, and to develop USP reference materials to aid FCC users in carrying out monograph tests. Reason to Participate: Participation in this project is an opportunity for leading color additive manufactures to help establish the high-bar of quality in FCC monograph standards. Table 1: Food Color Additives of Interest Allura Red AC Azorubine Patent Blue V Amaranth Erythrosine Ponceau 4R Brilliant Black PN Fast Green FCF Quinoline Yellow Brilliant Blue FCF Green S Red 2G Brown FK Indigotine Sunset Yellow FCF Brown HT Lithol Rubine BK Tartrazine Survey Instructions: For each synthetic food color additive that you produce, please provide as much information on the attached questions as possible to the Jeff Moore at FCC/USP. Jeff Moore, Ph.D. -

Investigation on the Presence of Sunset Yellow and Tartrazine in Commercial Beverages and Quantitation Using Ion-Pair Formation and Extraction
Investigation On The Presence of Sunset Yellow and Tartrazine In Commercial Beverages and Quantitation Using Ion-Pair Formation and Extraction Zailuddin Ariffin Faridah Hanum Hj. Badrun Baharudin Salleh In this work, 18 different commercial beverages were analyzed for the detection of Sunset Yellow and Tartrazine by paper chromatography. Quantitation of samples containing only one added colour proceeded using the ion pair formation and extraction method. The results obtained showed that all the commercial beverages tested contain synthetic colours permitted for use based on the Malaysian Food Act 1983. Quantitation of soft drinks which contained only one added colour complied with the U.K. standard, although one sample showed a slight excess compared to the set limit of 50 mg/L. Introduction Sunset Yellow (otherwise known as E110 or FD&C Yellow No. 6) and Tartrazine (also known as E102 or FD&C Yellow No. 5) are synthetic dyes (Fig. 1). They are commercially used as additives in pharmaceuticals and cosmetics, with the advantages that they can be easily mixed to achieve ideal colours and because of their low price compared to the natural dyes. Synthetic colourants normally contain azo functional groups and aromatic ring structures, so they are harmful to human health (6). For this reason, the controlled use and the accurate analysis of their contents in alimentary products is important. Fig. 1: Structures of Sunset Yellow and Tartrazine Sunset Yellow (FD&C Yellow No. 6) SO3Na SO3Na N N HO Tartrazine - + SO3 Na H O - + O3S Na N N N N + - H O2C Sunset Yellow is an azo food dye that was first used in 1929. -

Development and Validation of a Simple and Fast Method For
IOSR Journal of Environmental Science, Toxicology and Food Technology (IOSR-JESTFT) e-ISSN: 2319-2402,p- ISSN: 2319-2399.Volume 10, Issue 9 Ver. I (Sep. 2016), PP 17-22 www.iosrjournals.org Development and validation of a simple and fast method for simultaneous determination of sunset yellow and azorubine in carbonated orange flavor soft drinks samples by high- performance liquid chromatography Mohammad Faraji1, Gholamrezza Ghasempour1, Banafshe Nasiri Sahneh1, Rika Javanshir1, Mina Alavi 1Faculty of Food Industry and Agriculture, Department of Food science & Technology, Standard Research Institute (SRI), Karaj P.O. Box 31745-139, Iran Abstract: An efficient method was developed for the simultaneous determination of sunset yellow and azorubine in carbonated orange flavor soft drink samples by high performance liquid chromatography (HPLC- UV-Vis). Affecting parameters on separation and detection of the dyes were investigated and optimized. For these dyes good linearity (0.25–50 mg L-1, > r2=0.99) were obtained. Limits of detection for both of the dyes were 0.1 mg L-1. The recoveries of the dyes ranged from 92 to 102%. Intra and inter-day precision expressed as relative standard deviation (RSD%) at 5.0 and 50 mg L-1 levels less than 8.0% were also achieved. This method has been applied successfully in the determination of the sunset yellow and azorubine in soft drink samples. The average sunset yellow concentration of the 42 soft drink samples range between 20.6- 60.2 mg L-1. Also, azorubine was found in the soft drink samples in the range of N.D-4.4 mg L-1. -

Technological Applications of Natural Colorants in Food Systems: a Review
foods Review Technological Applications of Natural Colorants in Food Systems: A Review Ivan Luzardo-Ocampo 1 , Aurea K. Ramírez-Jiménez 2 , Jimena Yañez 2 , Luis Mojica 3 and Diego A. Luna-Vital 2,* 1 Instituto de Neurobiología, Universidad Nacional Autónoma de México (UNAM), Santiago de Querétaro, QRO 76230, Mexico; [email protected] 2 Tecnologico de Monterrey, School of Engineering and Science, Avenida Eugenio Garza Sada 2501 Sur, Monterrey, N. L. 64849, Mexico; [email protected] (A.K.R.-J.); [email protected] (J.Y.) 3 Tecnología Alimentaria, Centro de Investigación y Asistencia en Tecnología y Diseño del Estado de Jalisco (CIATEJ), A. C., Camino Arenero #1227 Col. El Bajío, Zapopan, JAL 45019, Mexico; [email protected] * Correspondence: [email protected] Abstract: Natural colorants have emerged as an alternative to their synthetic counterparts due to an existing health concern of these later. Moreover, natural-food colorants are a renewable option providing health benefits and interesting technological and sensory attributes to the food systems containing them. Several sources of natural colorants have been explored aiming to deliver the required wide color range demanded by consumers. This review aimed to compare and discuss the technological applications of the main natural-food colorants into food system in the last six years, giving additional information about their extraction process. Although natural colorants are promising choices to replace synthetic ones, optimization of processing conditions, research on new sources, and new formulations to ensure stability are required to equate their properties to their Citation: Luzardo-Ocampo, I.; synthetic counterparts. Ramírez-Jiménez, A.K.; Yañez, J.; Mojica, L.; Luna-Vital, D.A. -
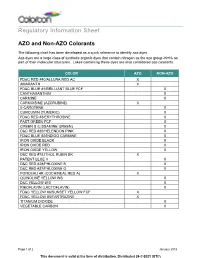
Regulatory Information Sheet AZO and Non-AZO Colorants
Regulatory Information Sheet AZO and Non-AZO Colorants The following chart has been developed as a quick reference to identify azo dyes. Azo dyes are a large class of synthetic organic dyes that contain nitrogen as the azo group -N=N- as part of their molecular structures. Lakes containing these dyes are also considered azo colorants. COLOR AZO NON-AZO FD&C RED #40/ALLURA RED AC X AMARANTH X FD&C BLUE #1/BRILLIANT BLUE FCF X CANTHAXANTHIN X CARMINE X CARMOISINE (AZORUBINE) X ß-CAROTENE X CURCUMIN (TUMERIC) X FD&C RED #3/ERYTHROSINE X FAST GREEN FCF X GREEN S (LISSAMINE GREEN) X D&C RED #30/HELENDON PINK X FD&C BLUE #2/INDIGO CARMINE X IRON OXIDE BLACK X IRON OXIDE RED X IRON OXIDE YELLOW X D&C RED #7/LITHOL RUBIN BK X PATENT BLUE V X D&C RED #28/PHLOXINE B X D&C RED #27/PHLOXINE O X PONCEAU 4R (COCHINEAL RED A) X QUINOLINE YELLOW WS X D&C YELLOW #10 X RIBOFLAVIN (LACTOFLAVIN) X FD&C YELLOW #6/SUNSET YELLOW FCF X FD&C YELLOW #5/TARTRAZINE X TITANIUM DIOXIDE X VEGETABLE CARBON X Page 1 of 2 January 2018 This document is valid at the time of distribution. Distributed 24-?-2021 (UTC) The information contained herein, to the best of our knowledge is true and accurate. Any recommendations or suggestions are made without warranty or guarantee, since the conditions of use are beyond our control. Any information contained herein is intended as a recommendation for use of our products so as not to infringe on any patent.