Neuromuscular Junction As an Entity of Nerve-Muscle Communication
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cell Biology and Fundamentals in Histology - En-Cours-2018-Liepr1004 Liepr1004 Cell Biology and Fundamentals in 2018 Histology
Université catholique de Louvain - Cell biology and fundamentals in histology - en-cours-2018-liepr1004 liepr1004 Cell biology and fundamentals in 2018 histology 5 credits 45.0 h Q2 Teacher(s) Behets Wydemans Catherine ;Henriet Patrick ; Language : French Place of the course Louvain-la-Neuve Main themes The major themes are : - Characteristics common to all living species - The human cell, its functioning and division - Classical, evolutive and molecular genetics - Cellular bases in sexual reproduction - The differents cell types and their organisation in tissues - The major steps in human embryonic development Aims By the end of the module, students should understand the bases of unicity and diversity in the living world. They will know the structure and functioning of human cell and genome as well as the mechanisms of 1 cell division and embryonic development. Moreover, they will know the structure of the major types of human tissues. - - - - The contribution of this Teaching Unit to the development and command of the skills and learning outcomes of the programme(s) can be accessed at the end of this sheet, in the section entitled “Programmes/courses offering this Teaching Unit”. Content (auteurs - titulaires actuels) : P. Henriet and Ph. van den Bosch de Aguilar 1. UNICITY IN THE LIVING WORLD 2. THE HUMAN CELL 3. DIVERSITY IN THE LIVING WORLD 4. MOLECULAR GENETICS 5. CELL DIVISION 6. GAMETOGENESIS AND FERTILIZATION 7. INTRODUCTION TO HUMAN EMBRYOLOGY Histology 1. EPITHELIAL TISSUE 2. CONNECTIVE TISSUE 3. BLOOD TISSUE 4. MUSCLE TISSUE -

Neural Control of Movement: Motor Neuron Subtypes, Proprioception and Recurrent Inhibition
List of Papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. I Enjin A, Rabe N, Nakanishi ST, Vallstedt A, Gezelius H, Mem- ic F, Lind M, Hjalt T, Tourtellotte WG, Bruder C, Eichele G, Whelan PJ, Kullander K (2010) Identification of novel spinal cholinergic genetic subtypes disclose Chodl and Pitx2 as mark- ers for fast motor neurons and partition cells. J Comp Neurol 518:2284-2304. II Wootz H, Enjin A, Wallen-Mackenzie Å, Lindholm D, Kul- lander K (2010) Reduced VGLUT2 expression increases motor neuron viability in Sod1G93A mice. Neurobiol Dis 37:58-66 III Enjin A, Leao KE, Mikulovic S, Le Merre P, Tourtellotte WG, Kullander K. 5-ht1d marks gamma motor neurons and regulates development of sensorimotor connections Manuscript IV Enjin A, Leao KE, Eriksson A, Larhammar M, Gezelius H, Lamotte d’Incamps B, Nagaraja C, Kullander K. Development of spinal motor circuits in the absence of VIAAT-mediated Renshaw cell signaling Manuscript Reprints were made with permission from the respective publishers. Cover illustration Carousel by Sasha Svensson Contents Introduction.....................................................................................................9 Background...................................................................................................11 Neural control of movement.....................................................................11 The motor neuron.....................................................................................12 Organization -

Immune Response and Histology of Humoral Rejection in Kidney
Document downloaded from http://www.elsevier.es, day 23/05/2017. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. n e f r o l o g i a 2 0 1 6;3 6(4):354–367 Revista de la Sociedad Española de Nefrología www.revistanefrologia.com Review Immune response and histology of humoral rejection in kidney transplantation a,∗ a b a Miguel González-Molina , Pedro Ruiz-Esteban , Abelardo Caballero , Dolores Burgos , a c a a Mercedes Cabello , Miriam Leon , Laura Fuentes , Domingo Hernandez a Nephrology Department, Regional University Hospital of Malaga, Malaga University, IBIMA, REDINREN RD12/0021/0015, Malaga, Spain b Immunology Department, Regional University Hospital of Malaga, Malaga University, IBIMA, REDINREN RD12/0021/0015, Malaga, Spain c Pathology Department, Regional University Hospital of Malaga, Malaga University, IBIMA, REDINREN RD12/0021/0015, Malaga, Spain a r t i c l e i n f o a b s t r a c t Article history: The adaptive immune response forms the basis of allograft rejection. Its weapons are direct Received 4 June 2015 cellular cytotoxicity, identified from the beginning of organ transplantation, and/or anti- Accepted 26 March 2016 bodies, limited to hyperacute rejection by preformed antibodies and not as an allogenic Available online 3 June 2016 response. This resulted in allogenic response being thought for decades to have just a cellu- lar origin. But the experimental studies by Gorer demonstrating tissue damage in allografts Keywords: due to antibodies secreted by B lymphocytes activated against polymorphic molecules were Immune response disregarded. -

ALS and Other Motor Neuron Diseases Can Represent Diagnostic Challenges
Review Article Address correspondence to Dr Ezgi Tiryaki, Hennepin ALS and Other Motor County Medical Center, Department of Neurology, 701 Park Avenue P5-200, Neuron Diseases Minneapolis, MN 55415, [email protected]. Ezgi Tiryaki, MD; Holli A. Horak, MD, FAAN Relationship Disclosure: Dr Tiryaki’s institution receives support from The ALS Association. Dr Horak’s ABSTRACT institution receives a grant from the Centers for Disease Purpose of Review: This review describes the most common motor neuron disease, Control and Prevention. ALS. It discusses the diagnosis and evaluation of ALS and the current understanding of its Unlabeled Use of pathophysiology, including new genetic underpinnings of the disease. This article also Products/Investigational covers other motor neuron diseases, reviews how to distinguish them from ALS, and Use Disclosure: Drs Tiryaki and Horak discuss discusses their pathophysiology. the unlabeled use of various Recent Findings: In this article, the spectrum of cognitive involvement in ALS, new concepts drugs for the symptomatic about protein synthesis pathology in the etiology of ALS, and new genetic associations will be management of ALS. * 2014, American Academy covered. This concept has changed over the past 3 to 4 years with the discovery of new of Neurology. genes and genetic processes that may trigger the disease. As of 2014, two-thirds of familial ALS and 10% of sporadic ALS can be explained by genetics. TAR DNA binding protein 43 kDa (TDP-43), for instance, has been shown to cause frontotemporal dementia as well as some cases of familial ALS, and is associated with frontotemporal dysfunction in ALS. Summary: The anterior horn cells control all voluntary movement: motor activity, res- piratory, speech, and swallowing functions are dependent upon signals from the anterior horn cells. -

Satellitosis, a Crosstalk Between Neurons, Vascular Structures and Neoplastic Cells in Brain Tumours; Early Manifestation of Invasive Behaviour
cancers Review Satellitosis, a Crosstalk between Neurons, Vascular Structures and Neoplastic Cells in Brain Tumours; Early Manifestation of Invasive Behaviour Prospero Civita 1,2,* , Ortenzi Valerio 3 , Antonio Giuseppe Naccarato 3 , Mark Gumbleton 2 and Geoffrey J. Pilkington 1,2,4,* 1 Brain Tumour Research Centre, Institute of Biological and Biomedical Sciences (IBBS), School of Pharmacy and Biomedical Sciences, University of Portsmouth, Portsmouth PO1 2DT, UK 2 School of Pharmacy and Pharmaceutical Sciences, College of Biomedical and Life Sciences, Cardiff University, Cardiff CF10 3NB, UK; gumbleton@cardiff.ac.uk 3 Department of Translational Research and New Technologies in Medicine and Surgery, Pisa University Hospital, 56100 Pisa, Italy; [email protected] (O.V.); [email protected] (A.G.N.) 4 Division of Neuroscience, Department of Basic and Clinical Neuroscience, Institute of Psychiatry & Neurology, King’s College London, London SE5 9RX, UK * Correspondence: CivitaP@cardiff.ac.uk (P.C.); geoff[email protected] (G.J.P.) Received: 9 November 2020; Accepted: 4 December 2020; Published: 11 December 2020 Simple Summary: This article reviews the concept of cellular satellitosis as originally described histologically by Santiago Ramón y Cajal in 1899 and Hans Joachim Scherer, more specifically in the context of glioblastoma invasiveness, during the early part of the 20th century. With the advent of new and emerging molecular technologies in the 21st century, the significance of both vascular and neuronal satellitosis by neoplastic cells offers intriguing possibilities into further clarifying the development, pathobiology and therapy of malignant glioma through closer investigation into the nature of these histological hallmarks. -

Basic Histology (23 Questions): Oral Histology (16 Questions
Board Question Breakdown (Anatomic Sciences section) The Anatomic Sciences portion of part I of the Dental Board exams consists of 100 test items. They are broken up into the following distribution: Gross Anatomy (50 questions): Head - 28 questions broken down in this fashion: - Oral cavity - 6 questions - Extraoral structures - 12 questions - Osteology - 6 questions - TMJ and muscles of mastication - 4 questions Neck - 5 questions Upper Limb - 3 questions Thoracic cavity - 5 questions Abdominopelvic cavity - 2 questions Neuroanatomy (CNS, ANS +) - 7 questions Basic Histology (23 questions): Ultrastructure (cell organelles) - 4 questions Basic tissues - 4 questions Bone, cartilage & joints - 3 questions Lymphatic & circulatory systems - 3 questions Endocrine system - 2 questions Respiratory system - 1 question Gastrointestinal system - 3 questions Genitouirinary systems - (reproductive & urinary) 2 questions Integument - 1 question Oral Histology (16 questions): Tooth & supporting structures - 9 questions Soft oral tissues (including dentin) - 5 questions Temporomandibular joint - 2 questions Developmental Biology (11 questions): Osteogenesis (bone formation) - 2 questions Tooth development, eruption & movement - 4 questions General embryology - 2 questions 2 National Board Part 1: Review questions for histology/oral histology (Answers follow at the end) 1. Normally most of the circulating white blood cells are a. basophilic leukocytes b. monocytes c. lymphocytes d. eosinophilic leukocytes e. neutrophilic leukocytes 2. Blood platelets are products of a. osteoclasts b. basophils c. red blood cells d. plasma cells e. megakaryocytes 3. Bacteria are frequently ingested by a. neutrophilic leukocytes b. basophilic leukocytes c. mast cells d. small lymphocytes e. fibrocytes 4. It is believed that worn out red cells are normally destroyed in the spleen by a. neutrophils b. -

A System for Studying Mechanisms of Neuromuscular Junction Development and Maintenance Valérie Vilmont1,‡, Bruno Cadot1, Gilles Ouanounou2 and Edgar R
© 2016. Published by The Company of Biologists Ltd | Development (2016) 143, 2464-2477 doi:10.1242/dev.130278 TECHNIQUES AND RESOURCES RESEARCH ARTICLE A system for studying mechanisms of neuromuscular junction development and maintenance Valérie Vilmont1,‡, Bruno Cadot1, Gilles Ouanounou2 and Edgar R. Gomes1,3,*,‡ ABSTRACT different animal models and cell lines (Chen et al., 2014; Corti et al., The neuromuscular junction (NMJ), a cellular synapse between a 2012; Lenzi et al., 2015) with the hope of recapitulating some motor neuron and a skeletal muscle fiber, enables the translation of features of neuromuscular diseases and understanding the triggers chemical cues into physical activity. The development of this special of one of their common hallmarks: the disruption of the structure has been subject to numerous investigations, but its neuromuscular junction (NMJ). The NMJ is one of the most complexity renders in vivo studies particularly difficult to perform. studied synapses. It is formed of three key elements: the lower motor In vitro modeling of the neuromuscular junction represents a powerful neuron (the pre-synaptic compartment), the skeletal muscle (the tool to delineate fully the fine tuning of events that lead to subcellular post-synaptic compartment) and the Schwann cell (Sanes and specialization at the pre-synaptic and post-synaptic sites. Here, we Lichtman, 1999). The NMJ is formed in a step-wise manner describe a novel heterologous co-culture in vitro method using rat following a series of cues involving these three cellular components spinal cord explants with dorsal root ganglia and murine primary and its role is basically to ensure the skeletal muscle functionality. -

The Histology of the Neuromuscular Junction In
75 THE HISTOLOGY OF THE NEUROMUSCULAR JUNCTION Downloaded from https://academic.oup.com/brain/article/84/1/75/372729 by guest on 27 September 2021 IN DYSTROPHIA MYOTONICA BY VIOLET MACDERMOT Department of Neurology, St. Thomas' Hospital, London, S.E.I (1) INTRODUCTION DYSTROPJHC MYOTONICA is a familial disease affecting males and females, usually presenting in adult life, characterized by muscular wasting and weakness together with certain other features. The muscles mainly in- volved are the temporal, masseter, facial, sternomastoid and limb muscles, in the latter those mainly affected being peripheral in distribution. A widespread disorder of muscular contraction, myotonia, is also present but is noticed chiefly in the tongue and in the muscles involved in grasping. The other features of the condition are some degree of mental defect, dysphonia, cataracts, frontal baldness, sparse body hair and testicular atrophy. Any of the manifestations of the disease may be absent and the order of presentation of symptoms is variable. The myotonia may precede muscular wasting by many years or may occur independently. In those muscles which are severely wasted the myotonia tends to disappear. The interest of dystrophia myotonica lies in the peculiar distribution of muscle involvement and in the combination of a disorder of muscle function with endocrine and other dysplasic features. The results of histological examination of biopsy and post-mortem material have been described and reviewed by numerous workers, notably Steinert (1909), Adie and Greenfield (1923), Keschner and Davison (1933), Hassin and Kesert (1948), Wohlfart (1951), Adams, Denny-Brown and Pearson (1953), Greenfield, Shy, Alvord and Berg (1957). -

Cranial Nerves 1, 5, 7-12
Cranial Nerve I Olfactory Nerve Nerve fiber modality: Special sensory afferent Cranial Nerves 1, 5, 7-12 Function: Olfaction Remarkable features: – Peripheral processes act as sensory receptors (the other special sensory nerves have separate Warren L Felton III, MD receptors) Professor and Associate Chair of Clinical – Primary afferent neurons undergo continuous Activities, Department of Neurology replacement throughout life Associate Professor of Ophthalmology – Primary afferent neurons synapse with secondary neurons in the olfactory bulb without synapsing Chair, Division of Neuro-Ophthalmology first in the thalamus (as do all other sensory VCU School of Medicine neurons) – Pathways to cortical areas are entirely ipsilateral 1 2 Crania Nerve I Cranial Nerve I Clinical Testing Pathology Anosmia, hyposmia: loss of or impaired Frequently overlooked in neurologic olfaction examination – 1% of population, 50% of population >60 years Aromatic stimulus placed under each – Note: patients with bilateral anosmia often report nostril with the other nostril occluded, eg impaired taste (ageusia, hypogeusia), though coffee, cloves, or soap taste is normal when tested Note that noxious stimuli such as Dysosmia: disordered olfaction ammonia are not used due to concomitant – Parosmia: distorted olfaction stimulation of CN V – Olfactory hallucination: presence of perceived odor in the absence of odor Quantitative clinical tests are available: • Aura preceding complex partial seizures of eg, University of Pennsylvania Smell temporal lobe origin -

Lancl1 Promotes Motor Neuron Survival and Extends the Lifespan of Amyotrophic Lateral Sclerosis Mice
Cell Death & Differentiation (2020) 27:1369–1382 https://doi.org/10.1038/s41418-019-0422-6 ARTICLE LanCL1 promotes motor neuron survival and extends the lifespan of amyotrophic lateral sclerosis mice 1 1 1 1 1 1 1 Honglin Tan ● Mina Chen ● Dejiang Pang ● Xiaoqiang Xia ● Chongyangzi Du ● Wanchun Yang ● Yiyuan Cui ● 1,2 1 1,3 4 4 3 1,5 Chao Huang ● Wanxiang Jiang ● Dandan Bi ● Chunyu Li ● Huifang Shang ● Paul F. Worley ● Bo Xiao Received: 19 November 2018 / Revised: 3 September 2019 / Accepted: 6 September 2019 / Published online: 30 September 2019 © The Author(s) 2019. This article is published with open access Abstract Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive loss of motor neurons. Improving neuronal survival in ALS remains a significant challenge. Previously, we identified Lanthionine synthetase C-like protein 1 (LanCL1) as a neuronal antioxidant defense gene, the genetic deletion of which causes apoptotic neurodegeneration in the brain. Here, we report in vivo data using the transgenic SOD1G93A mouse model of ALS indicating that CNS-specific expression of LanCL1 transgene extends lifespan, delays disease onset, decelerates symptomatic progression, and improves motor performance of SOD1G93A mice. Conversely, CNS-specific deletion of fl 1234567890();,: 1234567890();,: LanCL1 leads to neurodegenerative phenotypes, including motor neuron loss, neuroin ammation, and oxidative damage. Analysis reveals that LanCL1 is a positive regulator of AKT activity, and LanCL1 overexpression restores the impaired AKT activity in ALS model mice. These findings indicate that LanCL1 regulates neuronal survival through an alternative mechanism, and suggest a new therapeutic target in ALS. -

Quiescent Satellite Glial Cells of the Adult Trigeminal Ganglion
Cent. Eur. J. Med. • 9(3) • 2014 • 500-504 DOI: 10.2478/s11536-013-0285-z Central European Journal of Medicine Quiescent satellite glial cells of the adult trigeminal ganglion Research Article Mugurel C. Rusu*1,2,3, Valentina M. Mănoiu4, Nicolae Mirancea3, Gheorghe Nini5 1 „Carol Davila” University of Medicine and Pharmacy, 050511 Bucharest, Romania. 2 MEDCENTER - Center of Excellence in Laboratory Medicine and Pathology 013594 Bucharest, Romania 3 Institute of Biology of Bucharest – The Romanian Academy, , 060031 Bucharest, Romania 4 Faculty of Geography, University of Bucharest, 050107 Bucharest, Romania 5 Faculty of Medicine, Pharmacy and Dental Medicine, “Vasile Goldiş” Western University, 310045 Arad, Romania Received 18 August 2013; Accepted 27 November 2013 Abstract: Sensory ganglia comprise functional units built up by neurons and satellite glial cells (SGCs). In animal species there was proven the presence of neuronoglial progenitor cells in adult samples. Such neural crest-derived progenitors were found in immunohistochemistry (IHC). These fi ndings were not previously documented in transmission electron microscopy (TEM). It was thus aimed to assess in TEM if cells of the human adult trigeminal ganglion indeed have ultrastructural features to qualify for a progenitor, or quiescent phenotype. Trigeminal ganglia were obtained from fi fteen adult donor cadavers. In TEM, cells with heterochromatic nuclei, a pancytoplasmic content of free ribosomes, few perinuclear mitochondria, poor developed endoplasmic reticulum, lack of Golgi complexes and membrane traffi cking specializations, were found included in the neuronal envelopes built-up by SGCs. The ultrastructural pattern was strongly suggestive for these cells being quiescent progenitors. However, further experiments should correlate the morphologic and immune phenotypes of such cells. -
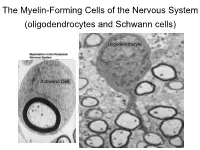
The Myelin-Forming Cells of the Nervous System (Oligodendrocytes and Schwann Cells)
The Myelin-Forming Cells of the Nervous System (oligodendrocytes and Schwann cells) Oligodendrocyte Schwann Cell Oligodendrocyte function Saltatory (jumping) nerve conduction Oligodendroglia PMD PMD Saltatory (jumping) nerve conduction Investigating the Myelinogenic Potential of Individual Oligodendrocytes In Vivo Sparse Labeling of Oligodendrocytes CNPase-GFP Variegated expression under the MBP-enhancer Cerebral Cortex Corpus Callosum Cerebral Cortex Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Ant Commissure Corpus Callosum Cerebral Cortex Caudate Putamen Piriform Cortex Corpus Callosum Ant Commissure Characterization of Oligodendrocyte Morphology Cerebral Cortex Corpus Callosum Caudate Putamen Cerebellum Brain Stem Spinal Cord Oligodendrocytes in disease: Cerebral Palsy ! CP major cause of chronic neurological morbidity and mortality in children ! CP incidence now about 3/1000 live births compared to 1/1000 in 1980 when we started intervening for ELBW ! Of all ELBW {gestation 6 mo, Wt. 0.5kg} , 10-15% develop CP ! Prematurely born children prone to white matter injury {WMI}, principle reason for the increase in incidence of CP ! ! 12 Cerebral Palsy Spectrum of white matter injury ! ! Macro Cystic Micro Cystic Gliotic Khwaja and Volpe 2009 13 Rationale for Repair/Remyelination in Multiple Sclerosis Oligodendrocyte specification oligodendrocytes specified from the pMN after MNs - a ventral source of oligodendrocytes