Molecular and Structural Requirements of the Β1l
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Properties and Units in Clinical Pharmacology and Toxicology
Pure Appl. Chem., Vol. 72, No. 3, pp. 479–552, 2000. © 2000 IUPAC INTERNATIONAL FEDERATION OF CLINICAL CHEMISTRY AND LABORATORY MEDICINE SCIENTIFIC DIVISION COMMITTEE ON NOMENCLATURE, PROPERTIES, AND UNITS (C-NPU)# and INTERNATIONAL UNION OF PURE AND APPLIED CHEMISTRY CHEMISTRY AND HUMAN HEALTH DIVISION CLINICAL CHEMISTRY SECTION COMMISSION ON NOMENCLATURE, PROPERTIES, AND UNITS (C-NPU)§ PROPERTIES AND UNITS IN THE CLINICAL LABORATORY SCIENCES PART XII. PROPERTIES AND UNITS IN CLINICAL PHARMACOLOGY AND TOXICOLOGY (Technical Report) (IFCC–IUPAC 1999) Prepared for publication by HENRIK OLESEN1, DAVID COWAN2, RAFAEL DE LA TORRE3 , IVAN BRUUNSHUUS1, MORTEN ROHDE1, and DESMOND KENNY4 1Office of Laboratory Informatics, Copenhagen University Hospital (Rigshospitalet), Copenhagen, Denmark; 2Drug Control Centre, London University, King’s College, London, UK; 3IMIM, Dr. Aiguader 80, Barcelona, Spain; 4Dept. of Clinical Biochemistry, Our Lady’s Hospital for Sick Children, Crumlin, Dublin 12, Ireland #§The combined Memberships of the Committee and the Commission (C-NPU) during the preparation of this report (1994–1996) were as follows: Chairman: H. Olesen (Denmark, 1989–1995); D. Kenny (Ireland, 1996); Members: X. Fuentes-Arderiu (Spain, 1991–1997); J. G. Hill (Canada, 1987–1997); D. Kenny (Ireland, 1994–1997); H. Olesen (Denmark, 1985–1995); P. L. Storring (UK, 1989–1995); P. Soares de Araujo (Brazil, 1994–1997); R. Dybkær (Denmark, 1996–1997); C. McDonald (USA, 1996–1997). Please forward comments to: H. Olesen, Office of Laboratory Informatics 76-6-1, Copenhagen University Hospital (Rigshospitalet), 9 Blegdamsvej, DK-2100 Copenhagen, Denmark. E-mail: [email protected] Republication or reproduction of this report or its storage and/or dissemination by electronic means is permitted without the need for formal IUPAC permission on condition that an acknowledgment, with full reference to the source, along with use of the copyright symbol ©, the name IUPAC, and the year of publication, are prominently visible. -

The Role of the Serotonergic System and the Effects of Antidepressants During Brain Development Examined Using in Vivo Pet Imaging and in Vitro Receptor Binding
From THE DEPARTMENT OF CLINICAL NEUROSCIENCE Karolinska Institutet, Stockholm, Sweden THE ROLE OF THE SEROTONERGIC SYSTEM AND THE EFFECTS OF ANTIDEPRESSANTS DURING BRAIN DEVELOPMENT EXAMINED USING IN VIVO PET IMAGING AND IN VITRO RECEPTOR BINDING Stal Saurav Shrestha Stockholm 2014 Cover Illustration: Voxel-wise analysis of the whole monkey brain using the PET radioligand, [11C]DASB showing persistent serotonin transporter upregulation even after more than 1.5 years of fluoxetine discontinuation. All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by Universitetsservice-AB © Stal Saurav Shrestha, 2014 ISBN 978-91-7549-522-4 Serotonergic System and Antidepressants During Brain Development To my family Amaze yourself ! Stal Saurav Shrestha, 2014 The Department of Clinical Neuroscience The role of the serotonergic system and the effects of antidepressants during brain development examined using in vivo PET imaging and in vitro receptor binding AKADEMISK AVHANDLING som för avläggande av medicine doktorsexamen vid Karolinska Institutet offentligen försvaras i CMM föreläsningssalen L8:00, Karolinska Universitetssjukhuset, Solna THESIS FOR DOCTORAL DEGREE (PhD) Stal Saurav Shrestha Date: March 31, 2014 (Monday); Time: 10 AM Venue: Center for Molecular Medicine Lecture Hall Floor 1, Karolinska Hospital, Solna Principal Supervisor: Opponent: Robert B. Innis, MD, PhD Klaus-Peter Lesch, MD, PhD National Institutes of Health University of Würzburg Department of NIMH Department -

33-Adrenoceptor-Mediated Relaxation Induced by Isoprenaline And
J. Smooth Muscle Res. 33 : 99-106. 99 The )32 and [33-Adrenoceptor-Mediated Relaxation Induced by Isoprenaline and Salbutamol in Guinea Pig Taenia Caecum Katsuo KOIKE, Tsukasa IcHiNo, Takahiro HORINOUCHI and Issei TAKAYANAGI Departmentof Chemical Pharmacology, Toho University School of PharmaceuticalSciences, 2-2-1, Miyama,Funabashi, Chiba 274, Japan Abstract To understand the receptor subtypes responsible for /3adrenoceptormediated relaxa tion of guinea pig taenia caecum, we investigated the effects of isoprenaline and salbutamol . Isoprenaline and salbutamol caused dose-dependent relaxation of the guinea pig taenia caecum. Propranolol, bupranolol and butoxamine produced shifts of the concentration response curves for isoprenaline and salbutamol. Schild regression analyses carried out for propranolol against isoprenaline and salbutamol gave pA2 values of 8.43 and 8.88, respective ly. Schild regression analyses carried out for butoxamine against isoprenaline and sal butamol gave pA2 values of 6.46 and 6.68, respectively. Schild regression analyses carried out for bupranolol against isoprenaline and salbutamol gave pA2 values of 8.60 and 8.69, respectively. However, in the presence of 3 x 10' M atenolol, 10-4 M butoxamine and 10-6 M phentolamine to block the fir , /32 and a -adrenoceptor effects, respectively, Schild regression analyses carried out for bupranolol against isoprenaline and salbutamol gave pA2 values of 5.77 and 5.97, respectively. These results suggest that the relaxant responses to isoprenaline and salbutamol in the guinea pig taenia caecum are mediated by both the /.32 and the A-adrenoceptors. Key words : f2-adrenoceptor, A-adrenoceptor, isoprenaline, salbutamol , guinea pig taenia caecum Introduction The /3adrenoceptors were subclassified as and /32subtypes based on the agonist potency and tissue localization. -
Guidance for the Format and Content of the Protocol of Non-Interventional
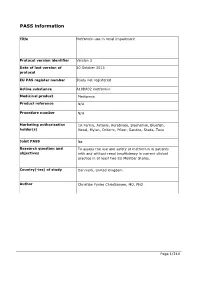
PASS information Title Metformin use in renal impairment Protocol version identifier Version 2 Date of last version of 30 October 2013 protocol EU PAS register number Study not registered Active substance A10BA02 metformin Medicinal product Metformin Product reference N/A Procedure number N/A Marketing authorisation 1A Farma, Actavis, Aurobindo, Biochemie, Bluefish, holder(s) Hexal, Mylan, Orifarm, Pfizer, Sandoz, Stada, Teva Joint PASS No Research question and To assess the use and safety of metformin in patients objectives with and without renal insufficiency in current clinical practice in at least two EU Member States. Country(-ies) of study Denmark, United Kingdom Author Christian Fynbo Christiansen, MD, PhD Page 1/214 Marketing authorisation holder(s) Marketing authorisation N/A holder(s) MAH contact person N/A Page 2/214 1. Table of Contents PASS information .......................................................................................................... 1 Marketing authorisation holder(s) .................................................................................... 2 1. Table of Contents ...................................................................................................... 3 2. List of abbreviations ................................................................................................... 4 3. Responsible parties .................................................................................................... 5 4. Abstract .................................................................................................................. -

Data Supplements
Perioperative Beta Blockade in Noncardiac Surgery: A Systematic Review for the 2014 ACC/AHA Guideline on Perioperative Cardiovascular Evaluation and Management of Patients Undergoing Noncardiac Surgery Data Supplements Data Supplement 1. Electronic Search Terms Within PubMed Database ..................................................................................................................... 2 Data Supplement 2. Electronic Search Terms Within Embase Database ...................................................................................................................... 3 Data Supplement 3. Electronic Search Terms Within Cochrane Central Register of Controlled Trials ....................................................................... 4 Data Supplement 4. Relevance, Fidelity, and Risk of Bias* of Included RCTs ............................................................................................................ 5 Data Supplement 5. Quality and Results of Included Observational Studies ................................................................................................................ 6 Data Supplement 6. Definitions of Significant Perioperative Hypotension and Bradycardia in Included RCTs .......................................................... 6 Data Supplement 7. Flow Diagram Describing Literature Search Results for the Systematic Review ......................................................................... 8 Data Supplement 8. Effect of Perioperative Beta Blockade on In-Hospital or 30-Day Cardiovascular -

Novel Method of Administering Β-Blockers and Novel Dosage Forms Containing Same Anwar A
University of Kentucky UKnowledge Pharmaceutical Sciences Faculty Patents Pharmaceutical Sciences 1-31-1984 Novel Method of Administering β-Blockers and Novel Dosage Forms Containing Same Anwar A. Hussain University of Kentucky Right click to open a feedback form in a new tab to let us know how this document benefits oy u. Follow this and additional works at: https://uknowledge.uky.edu/ps_patents Part of the Pharmacy and Pharmaceutical Sciences Commons Recommended Citation Hussain, Anwar A., "Novel Method of Administering β-Blockers and Novel Dosage Forms Containing Same" (1984). Pharmaceutical Sciences Faculty Patents. 124. https://uknowledge.uky.edu/ps_patents/124 This Patent is brought to you for free and open access by the Pharmaceutical Sciences at UKnowledge. It has been accepted for inclusion in Pharmaceutical Sciences Faculty Patents by an authorized administrator of UKnowledge. For more information, please contact [email protected]. Unlted States Patent [191 [11] 4,428,883 Hussain [45] Jan. 31, 1984 [54] NOVEL METHOD OF ADMINISTERING 4,012,444 3/l977 Lunts ................................. .. 424/ 324 B-BLOCKERSF0 RMS Co NTA' AND, INING NOVEL SAME DOSAGE ' 4,250,163, , 1.1’;2/l981 llieoll’loldNagaioe a ......................................... ---- -~.. .. 424/14 [7 5 ] I nventor : An war A . Hussam,° Lexington' , Ky . FOREIGN PATENT DOCUMENTS [73] Assignee: The University of Kentucky Research . Foundation’ Lexington, Ky. 979389 1/ 1965 United Klngdom . I [21] APPL Nod 241,413 OTHER PUBLICATIONS . Stern, Arzmit. Forsch, vol. 24, 1974, pp. 70-71. [22] Flled‘ Mm‘- 6’ 1981 Black, Brit. J. PharmacoL, (1965) 25, pp. 577-591. [51] Int. Cl.3 ................... .. A61K 27/00; A61K 31/15; Prima'y Examiner_stanley L Friedman A61K 31/40; A61K 31/47; A61K 31/135; Attorney, Agent, or Firm-Burns, Doane, Swecker & A61K 31/165; A61K 31/435; A61K 31/475; Mathis A61K 47/00; C07C 143/90; CllD 1/28; C09F - 5 /()() [57] ABSTRACT [52] US. -
![Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set](https://docslib.b-cdn.net/cover/8870/ehealth-dsi-ehdsi-v2-2-2-or-ehealth-dsi-master-value-set-1028870.webp)
Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set
MTC eHealth DSI [eHDSI v2.2.2-OR] eHealth DSI – Master Value Set Catalogue Responsible : eHDSI Solution Provider PublishDate : Wed Nov 08 16:16:10 CET 2017 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 1 of 490 MTC Table of Contents epSOSActiveIngredient 4 epSOSAdministrativeGender 148 epSOSAdverseEventType 149 epSOSAllergenNoDrugs 150 epSOSBloodGroup 155 epSOSBloodPressure 156 epSOSCodeNoMedication 157 epSOSCodeProb 158 epSOSConfidentiality 159 epSOSCountry 160 epSOSDisplayLabel 167 epSOSDocumentCode 170 epSOSDoseForm 171 epSOSHealthcareProfessionalRoles 184 epSOSIllnessesandDisorders 186 epSOSLanguage 448 epSOSMedicalDevices 458 epSOSNullFavor 461 epSOSPackage 462 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 2 of 490 MTC epSOSPersonalRelationship 464 epSOSPregnancyInformation 466 epSOSProcedures 467 epSOSReactionAllergy 470 epSOSResolutionOutcome 472 epSOSRoleClass 473 epSOSRouteofAdministration 474 epSOSSections 477 epSOSSeverity 478 epSOSSocialHistory 479 epSOSStatusCode 480 epSOSSubstitutionCode 481 epSOSTelecomAddress 482 epSOSTimingEvent 483 epSOSUnits 484 epSOSUnknownInformation 487 epSOSVaccine 488 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 3 of 490 MTC epSOSActiveIngredient epSOSActiveIngredient Value Set ID 1.3.6.1.4.1.12559.11.10.1.3.1.42.24 TRANSLATIONS Code System ID Code System Version Concept Code Description (FSN) 2.16.840.1.113883.6.73 2017-01 A ALIMENTARY TRACT AND METABOLISM 2.16.840.1.113883.6.73 2017-01 -

(12) United States Patent (10) Patent No.: US 6,258,350 B1 Mallick (45) Date of Patent: Jul
USOO625.8350B1 (12) United States Patent (10) Patent No.: US 6,258,350 B1 Mallick (45) Date of Patent: Jul. 10, 2001 (54) SUSTAINED RELEASE OPHTHALMIC Stalker et al., “Enhancement of Ocular Drug Bioavailability FORMULATION Through the Use of Drug-Resin Complexes II: Aqueous Humor Concentrations Versus Time Profiles for Nafcillin,” (75) Inventor: Sushanta Mallick, Dallas, TX (US) Abstract of Papers Presented Before the American Pharma ceutical ASSociation Academy of Pharmaceutical Sciences (73) Assignee: Alcon Manufacturing, Ltd., Fort 33' National Meeting, San Diego, CA: vol. 12 (2), p. 116, Worth, TX (US) No. 58 (1982). Rohm and Hass Product Brochure for Amberlite(E) and (*) Notice: Subject to any disclaimer, the term of this Duolite(R) (1991). patent is extended or adjusted under 35 Schoenwald et al., “Influence of High-Viscosity Vehicles on U.S.C. 154(b) by 0 days. Miotic Effect of Pilocarpine,” J. of Pharm. Sciences, vol. 67(9), pp. 1280–1283 (1978). (21) Appl. No.: 09/456,617 Stalker “Enhancement of Ocular Drug Bioavailability Through the Use of Micronized, Functionalized Polymers as (22) Filed: Dec. 8, 1999 Carriers of Therapeutic Agents,” Dissertation Submitted at the University of Kentucky (1983). Related U.S. Application Data Machin et al., "B-Selective Adrenoceptor Antagonists. 2. (60) Provisional application No. 60/116,487, filed on Jan. 20, 4-Ether-Linker Phenoxypropanolamines,” J.Med. Chem., 1999. vol. 26, pp. 1570–1576 (1983). McClure et al., “Antihypertensive -3-Adrenergic Blocking (51) Int. Cl. ............................ A61K 9/10; A61K 47/32 Agents: N-Aralkyl analogues of 2-3-(tert-Butylamino)-2-hydroxypropoxy-3-cyanopyri (52) U.S. -

The 2.1 A˚Resolution Structure of Cyanopindolol-Bound B1
The 2.1 A˚ Resolution Structure of Cyanopindolol-Bound + b1-Adrenoceptor Identifies an Intramembrane Na Ion that Stabilises the Ligand-Free Receptor Jennifer L. Miller-Gallacher.¤a, Rony Nehme´ ., Tony Warne, Patricia C. Edwards, Gebhard F. X. Schertler¤b¤c, Andrew G. W. Leslie, Christopher G. Tate* Structural Studies Division, MRC Laboratory of Molecular Biology, Cambridge, Cambridgeshire, United Kingdom Abstract The b1-adrenoceptor (b1AR) is a G protein-coupled receptor (GPCR) that is activated by the endogenous agonists adrenaline and noradrenaline. We have determined the structure of an ultra-thermostable b1AR mutant bound to the weak partial agonist cyanopindolol to 2.1 A˚ resolution. High-quality crystals (100 mm plates) were grown in lipidic cubic phase without the assistance of a T4 lysozyme or BRIL fusion in cytoplasmic loop 3, which is commonly employed for GPCR crystallisation. An intramembrane Na+ ion was identified co-ordinated to Asp872.50, Ser1283.39 and 3 water molecules, which is part of a more extensive network of water molecules in a cavity formed between transmembrane helices 1, 2, 3, 6 and 7. Remarkably, + this water network and Na ion is highly conserved between b1AR and the adenosine A2A receptor (rmsd of 0.3 A˚), despite an overall rmsd of 2.4 A˚ for all Ca atoms and only 23% amino acid identity in the transmembrane regions. The affinity of + agonist binding and nanobody Nb80 binding to b1AR is unaffected by Na ions, but the stability of the receptor is decreased by 7.5uC in the absence of Na+. Mutation of amino acid side chains that are involved in the co-ordination of either + Na or water molecules in the network decreases the stability of b1AR by 5–10uC. -

An Insight on Analytical Profile on Bisoprolol Fumarate – a Selective Beta-1 Adrenoreceptor Blocker
DOI: 10.15415/jptrm.2017.52012 An Insight on Analytical Profile on Bisoprolol Fumarate – A Selective Beta-1 Adrenoreceptor Blocker AJINKYA G. DHANDAR, SURAJ R. CHAUDHARI, SAURABH B. GANORKAR, AMOD S. PATIL, SANJAY J. SURANA AND ATUL A. SHIRKHEDKAR* R.C. Patel Institute of Pharmaceutical Education and Research, Shirpur, Dist. Dhule, India *Email: [email protected] Received: June 15, 2017 | Revised: July 19, 2017 | Accepted: Sept. 22, 2017 Published online: Nov. 02, 2017 The Author(s) 2017. This article is published with open access at www.chitkara.edu.in/publications Abstract BF is Beta-adreno receptor antagonist and used as an Anti- Hypertensive Drug. BF gives the blocking action on β1-adrenergic receptors in the heart and vascular smooth muscle. The present review compiles the various approaches implemented for quantification of BF in bulk drug, pharmaceutical matrix and biological fluid. This review represents more than 50 analytical methods which include capillary electrophoresis, HPLC, HPTLC, UV-Spectroscopy, UPLC, impurity profiling and electrochemical methods implemented for estimation of BF as a single component as well as in multicomponent. Keyword: BF; Bioanalytical; UPLC/LC-MS; capillary electrophoresis; impurity profile 1. INTRODUCTION BF is an extremely discriminatory β1-adrenergic blocker [1]. BF is chemically: (RS)-1-[4-[[2-(1- Methylethoxy) ethoxy] methyl] phenoxy] -3-[(1 methyl ethyl) amino] propan-2-ol fumarate Figure 1. It is official in, USP. BF has similar structure to metoprolol, bopindolol, hydrochlorothiazide, atenolol [2]. Structure of BF, there is two substituents β at para position of benzene provide the activity of -selectivity, In which Journal of Pharmaceutical it has two substituents in para position of benzene which might be the Technology, Research and activity of β- selectivity [3]. -

Adrenergic G-Protein- Coupled Receptor
Vol 454 | 24 July 2008 | doi:10.1038/nature07101 ARTICLES Structure of a b1-adrenergic G-protein- coupled receptor Tony Warne1, Maria J. Serrano-Vega1, Jillian G. Baker2, Rouslan Moukhametzianov1, Patricia C. Edwards1, Richard Henderson1, Andrew G. W. Leslie1, Christopher G. Tate1 & Gebhard F. X. Schertler1 G-protein-coupled receptors have a major role in transmembrane signalling in most eukaryotes and many are important drug targets. Here we report the 2.7 A˚ resolution crystal structure of a b1-adrenergic receptor in complex with the high-affinity antagonist cyanopindolol. The modified turkey (Meleagris gallopavo) receptor was selected to be in its antagonist conformation and its thermostability improved by earlier limited mutagenesis. The ligand-binding pocket comprises 15 side chains from amino acid residues in 4 transmembrane a-helices and extracellular loop 2. This loop defines the entrance of the ligand-binding pocket and is stabilized by two disulphide bonds and a sodium ion. Binding of cyanopindolol to the b1-adrenergic receptor and binding of carazolol to the b2-adrenergic receptor involve similar interactions. A short well-defined helix in cytoplasmic loop 2, not observed in either rhodopsin or the b2-adrenergic receptor, directly interacts by means of a tyrosine with the highly conserved DRY motif at the end of helix 3 that is essential for receptor activation. G-protein-coupled receptors (GPCRs) are a large family of integral These structures, both containing the high affinity antagonist cara- membrane proteins that are prevalent in eukaryotes from yeast to zolol, defined the overall architecture of b2AR and the structure of the man, and function as key intermediaries in the transduction of sig- ligand-binding pocket. -

Drugs for Primary Prevention of Atherosclerotic Cardiovascular Disease: an Overview of Systematic Reviews
Supplementary Online Content Karmali KN, Lloyd-Jones DM, Berendsen MA, et al. Drugs for primary prevention of atherosclerotic cardiovascular disease: an overview of systematic reviews. JAMA Cardiol. Published online April 27, 2016. doi:10.1001/jamacardio.2016.0218. eAppendix 1. Search Documentation Details eAppendix 2. Background, Methods, and Results of Systematic Review of Combination Drug Therapy to Evaluate for Potential Interaction of Effects eAppendix 3. PRISMA Flow Charts for Each Drug Class and Detailed Systematic Review Characteristics and Summary of Included Systematic Reviews and Meta-analyses eAppendix 4. List of Excluded Studies and Reasons for Exclusion This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. 1 Downloaded From: https://jamanetwork.com/ on 09/28/2021 eAppendix 1. Search Documentation Details. Database Organizing body Purpose Pros Cons Cochrane Cochrane Library in Database of all available -Curated by the Cochrane -Content is limited to Database of the United Kingdom systematic reviews and Collaboration reviews completed Systematic (UK) protocols published by by the Cochrane Reviews the Cochrane -Only systematic reviews Collaboration Collaboration and systematic review protocols Database of National Health Collection of structured -Curated by Centre for -Only provides Abstracts of Services (NHS) abstracts and Reviews and Dissemination structured abstracts Reviews of Centre for Reviews bibliographic