Identification of Monotonically Differentially Expressed Genes for Non-Small Cell Lung Cancer Suyan Tian
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Data
Figure 2S 4 7 A - C 080125 CSCs 080418 CSCs - + IFN-a 48 h + IFN-a 48 h + IFN-a 72 h 6 + IFN-a 72 h 3 5 MRFI 4 2 3 2 1 1 0 0 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 7 B 13 080125 FBS - D 080418 FBS - + IFN-a 48 h 12 + IFN-a 48 h + IFN-a 72 h + IFN-a 72 h 6 080125 FBS 11 10 5 9 8 4 7 6 3 MRFI 5 4 2 3 2 1 1 0 0 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 Molecule Molecule FIGURE 4S FIGURE 5S Panel A Panel B FIGURE 6S A B C D Supplemental Results Table 1S. Modulation by IFN-α of APM in GBM CSC and FBS tumor cell lines. Molecule * Cell line IFN-α‡ HLA β2-m# HLA LMP TAP1 TAP2 class II A A HC§ 2 7 10 080125 CSCs - 1∞ (1) 3 (65) 2 (91) 1 (2) 6 (47) 2 (61) 1 (3) 1 (2) 1 (3) + 2 (81) 11 (80) 13 (99) 1 (3) 8 (88) 4 (91) 1 (2) 1 (3) 2 (68) 080125 FBS - 2 (81) 4 (63) 4 (83) 1 (3) 6 (80) 3 (67) 2 (86) 1 (3) 2 (75) + 2 (99) 14 (90) 7 (97) 5 (75) 7 (100) 6 (98) 2 (90) 1 (4) 3 (87) 080418 CSCs - 2 (51) 1 (1) 1 (3) 2 (47) 2 (83) 2 (54) 1 (4) 1 (2) 1 (3) + 2 (81) 3 (76) 5 (75) 2 (50) 2 (83) 3 (71) 1 (3) 2 (87) 1 (2) 080418 FBS - 1 (3) 3 (70) 2 (88) 1 (4) 3 (87) 2 (76) 1 (3) 1 (3) 1 (2) + 2 (78) 7 (98) 5 (99) 2 (94) 5 (100) 3 (100) 1 (4) 2 (100) 1 (2) 070104 CSCs - 1 (2) 1 (3) 1 (3) 2 (78) 1 (3) 1 (2) 1 (3) 1 (3) 1 (2) + 2 (98) 8 (100) 10 (88) 4 (89) 3 (98) 3 (94) 1 (4) 2 (86) 2 (79) * expression of APM molecules was evaluated by intracellular staining and cytofluorimetric analysis; ‡ cells were treatead or not (+/-) for 72 h with 1000 IU/ml of IFN-α; # β-2 microglobulin; § β-2 microglobulin-free HLA-A heavy chain; ∞ values are indicated as ratio between the mean of fluorescence intensity of cells stained with the selected mAb and that of the negative control; bold values indicate significant MRFI (≥ 2). -

Genetic Analysis of Retinopathy in Type 1 Diabetes
Genetic Analysis of Retinopathy in Type 1 Diabetes by Sayed Mohsen Hosseini A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Institute of Medical Science University of Toronto © Copyright by S. Mohsen Hosseini 2014 Genetic Analysis of Retinopathy in Type 1 Diabetes Sayed Mohsen Hosseini Doctor of Philosophy Institute of Medical Science University of Toronto 2014 Abstract Diabetic retinopathy (DR) is a leading cause of blindness worldwide. Several lines of evidence suggest a genetic contribution to the risk of DR; however, no genetic variant has shown convincing association with DR in genome-wide association studies (GWAS). To identify common polymorphisms associated with DR, meta-GWAS were performed in three type 1 diabetes cohorts of White subjects: Diabetes Complications and Control Trial (DCCT, n=1304), Wisconsin Epidemiologic Study of Diabetic Retinopathy (WESDR, n=603) and Renin-Angiotensin System Study (RASS, n=239). Severe (SDR) and mild (MDR) retinopathy outcomes were defined based on repeated fundus photographs in each study graded for retinopathy severity on the Early Treatment Diabetic Retinopathy Study (ETDRS) scale. Multivariable models accounted for glycemia (measured by A1C), diabetes duration and other relevant covariates in the association analyses of additive genotypes with SDR and MDR. Fixed-effects meta- analysis was used to combine the results of GWAS performed separately in WESDR, ii RASS and subgroups of DCCT, defined by cohort and treatment group. Top association signals were prioritized for replication, based on previous supporting knowledge from the literature, followed by replication in three independent white T1D studies: Genesis-GeneDiab (n=502), Steno (n=936) and FinnDiane (n=2194). -

Deregulated Gene Expression Pathways in Myelodysplastic Syndrome Hematopoietic Stem Cells
Leukemia (2010) 24, 756–764 & 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 $32.00 www.nature.com/leu ORIGINAL ARTICLE Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells A Pellagatti1, M Cazzola2, A Giagounidis3, J Perry1, L Malcovati2, MG Della Porta2,MJa¨dersten4, S Killick5, A Verma6, CJ Norbury7, E Hellstro¨m-Lindberg4, JS Wainscoat1 and J Boultwood1 1LRF Molecular Haematology Unit, NDCLS, John Radcliffe Hospital, Oxford, UK; 2Department of Hematology Oncology, University of Pavia Medical School, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy; 3Medizinische Klinik II, St Johannes Hospital, Duisburg, Germany; 4Division of Hematology, Department of Medicine, Karolinska Institutet, Stockholm, Sweden; 5Department of Haematology, Royal Bournemouth Hospital, Bournemouth, UK; 6Albert Einstein College of Medicine, Bronx, NY, USA and 7Sir William Dunn School of Pathology, University of Oxford, Oxford, UK To gain insight into the molecular pathogenesis of the the World Health Organization.6,7 Patients with refractory myelodysplastic syndromes (MDS), we performed global gene anemia (RA) with or without ringed sideroblasts, according to expression profiling and pathway analysis on the hemato- poietic stem cells (HSC) of 183 MDS patients as compared with the the French–American–British classification, were subdivided HSC of 17 healthy controls. The most significantly deregulated based on the presence or absence of multilineage dysplasia. In pathways in MDS include interferon signaling, thrombopoietin addition, patients with RA with excess blasts (RAEB) were signaling and the Wnt pathways. Among the most signifi- subdivided into two categories, RAEB1 and RAEB2, based on the cantly deregulated gene pathways in early MDS are immuno- percentage of bone marrow blasts. -

Genomic Alterations of Ground-Glass Nodular Lung Adenocarcinoma
www.nature.com/scientificreports OPEN Genomic alterations of ground- glass nodular lung adenocarcinoma Hyun Lee1, Je-Gun Joung2, Hyun-Tae Shin2, Duk-Hwan Kim3, Yujin Kim3, Hojoong Kim1, O. Jung Kwon1, Young Mog Shim4, Ho Yun Lee5, Kyung Soo Lee5, Yoon-La Choi6, 2 7 1 Received: 1 February 2018 Woong-Yang Park , D. Neil Hayes & Sang-Won Um Accepted: 30 April 2018 In-depth molecular pathogenesis of ground-glass nodular lung adenocarcinoma has not been well Published: xx xx xxxx understood. The objectives of this study were to identify genomic alterations in ground-glass nodular lung adenocarcinomas and to investigate whether viral transcripts were detected in these tumors. Nine patients with pure (n = 4) and part-solid (n = 5) ground-glass nodular adenocarcinomas were included. Six were females with a median age of 58 years. We performed targeted exon sequencing and RNA sequencing. EGFR (n = 10), IDH2 (n = 2), TP53 (n = 1), PTEN (n = 1), EPHB4 (n = 1), and BRAF (n = 1) were identifed as driver mutations by targeted exon sequencing. Vasculogenesis-associated genes including NOTCH4 and TGFBR3 expression were signifcantly downregulated in adenocarcinoma tissue versus normal tissue (adjusted P values < 0.001 for both NOTCH4 and TGFBR3). In addition, fve novel fusion gene loci were identifed in four lung adenocarcinomas. However, no signifcant virus-associated transcripts were detected in tumors. In conclusions, EGFR, IDH2, TP53, PTEN, EPHB4, and BRAF were identifed as putative driver mutations of ground-glass nodular adenocarcinomas. Five novel fusion genes were also identifed in four tumors. Viruses do not appear to be involved in the tumorigenesis of ground-glass nodular lung adenocarcinoma. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Roles and Mechanisms of Kinesin-6 KIF20A in Spindle Organization During Cell Division T ⁎ Wen-Da Wu, Kai-Wei Yu, Ning Zhong, Yu Xiao, Zhen-Yu She
European Journal of Cell Biology 98 (2019) 74–80 Contents lists available at ScienceDirect European Journal of Cell Biology journal homepage: www.elsevier.com/locate/ejcb Review Roles and mechanisms of Kinesin-6 KIF20A in spindle organization during cell division T ⁎ Wen-Da Wu, Kai-Wei Yu, Ning Zhong, Yu Xiao, Zhen-Yu She Department of Cell Biology and Genetics/Center for Cell and Developmental Biology, The School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian 350108, China ARTICLE INFO ABSTRACT Keywords: Mitotic kinesin is crucial for spindle assembly and chromosome segregation in cell division. KIF20A/MKlp2, a Kinesin-6 member of kinesin-6 subfamily, plays important roles in the central spindle organization at anaphase and cy- KIF20A tokinesis. In this review, we briefly introduce the discovery and classification of kinesin-6 motors in model Microtubule organisms, and summarize the biochemical features and mechanics of KIF20A proteins. We emphasize the Anaphase complicated interactions of KIF20A with partner proteins, including MKlp1, Plk1 and Rab6. Particularly, we Spindle assembly highlight the regulation of Cdk1 and chromosomal passenger complex on kinesin-6 KIF20A at late stage of Mitosis mitosis. We summarized the multiple functions of KIF20A in central spindle assembly and the formation of cleavage furrow in both mitosis and meiosis. In addition, we conclude the expression patterns of KIF20A in tumorigenesis and its applications in tumor therapy. 1. Introduction kinesin superfamily proteins (Miki et al., 2005). Kinesin-6 subfamily is comprised of KIF20A (Lawrence et al., 2004), KIF20B (MPP1) Kinesin superfamily proteins (KIFs) are molecular motors that (Kamimoto et al., 2001; Matsumoto-Taniura et al., 1996; Westendorf mediate the transport of various cargos, including the newly synthe- et al., 1994) and MKlp1 (Lawrence et al., 2004; Nislow et al., 1990; sized protein complexes, vesicles and mRNAs along the microtubule Sellitto and Kuriyama, 1988). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
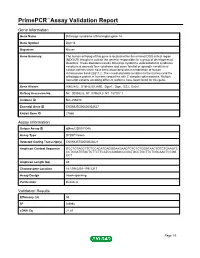
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name DiGeorge syndrome critical region gene 14 Gene Symbol Dgcr14 Organism Mouse Gene Summary The human ortholog of this gene is located within the minimal DGS critical region (MDGCR) thought to contain the gene(s) responsible for a group of developmental disorders. These disorders include DiGeorge syndrome velocardiofacial syndrome conotruncal anomaly face syndrome and some familial or sporadic conotruncal cardiac defects which have been associated with microdeletion of human chromosome band 22q11.2. The encoded protein localizes to the nucleus and the orthologous protein in humans co-purifies with C complex spliceosomes. Multiple transcript variants encoding different isoforms have been found for this gene. Gene Aliases AI462402, D16H22S1269E, Dgcr1, Dgsi, ES2, Es2el RefSeq Accession No. NC_000082.6, NT_039624.8, NT_187007.1 UniGene ID Mm.256480 Ensembl Gene ID ENSMUSG00000003527 Entrez Gene ID 27886 Assay Information Unique Assay ID qMmuCID0011048 Assay Type SYBR® Green Detected Coding Transcript(s) ENSMUST00000003621 Amplicon Context Sequence GCCTGTAGCTTCTCCACATCAGGGAAGAAGTCTCTCTGGATAACTGTCTGAAGTC CCTCGATGTACTCTTCTTCATCCAGGACCCGCTGCCTGCTTCTCGCAACTCCGG CCT Amplicon Length (bp) 82 Chromosome Location 16:17910201-17911217 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 95 R2 0.9994 cDNA Cq 21.87 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 79.5 gDNA Cq 23.94 Specificity (%) 100 Information to assist with data interpretation is provided -

CRNKL1 Is a Highly Selective Regulator of Intron-Retaining HIV-1 and Cellular Mrnas
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.04.934927; this version posted February 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 CRNKL1 is a highly selective regulator of intron-retaining HIV-1 and cellular mRNAs 2 3 4 Han Xiao1, Emanuel Wyler2#, Miha Milek2#, Bastian Grewe3, Philipp Kirchner4, Arif Ekici4, Ana Beatriz 5 Oliveira Villela Silva1, Doris Jungnickl1, Markus Landthaler2,5, Armin Ensser1, and Klaus Überla1* 6 7 1 Institute of Clinical and Molecular Virology, University Hospital Erlangen, Friedrich-Alexander 8 Universität Erlangen-Nürnberg, Erlangen, Germany 9 2 Berlin Institute for Medical Systems Biology, Max-Delbrück-Center for Molecular Medicine in the 10 Helmholtz Association, Robert-Rössle-Strasse 10, 13125, Berlin, Germany 11 3 Department of Molecular and Medical Virology, Ruhr-University, Bochum, Germany 12 4 Institute of Human Genetics, University Hospital Erlangen, Friedrich-Alexander Universität Erlangen- 13 Nürnberg, Erlangen, Germany 14 5 IRI Life Sciences, Institute für Biologie, Humboldt Universität zu Berlin, Philippstraße 13, 10115, Berlin, 15 Germany 16 # these two authors contributed equally 17 18 19 *Corresponding author: 20 Prof. Dr. Klaus Überla 21 Institute of Clinical and Molecular Virology, University Hospital Erlangen 22 Friedrich-Alexander Universität Erlangen-Nürnberg 23 Schlossgarten 4, 91054 Erlangen 24 Germany 25 Tel: (+49) 9131-8523563 26 e-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.02.04.934927; this version posted February 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Functional Characterization of the New 8Q21 Asthma Risk Locus
Functional characterization of the new 8q21 Asthma risk locus Cristina M T Vicente B.Sc, M.Sc A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2017 Faculty of Medicine Abstract Genome wide association studies (GWAS) provide a powerful tool to identify genetic variants associated with asthma risk. However, the target genes for many allergy risk variants discovered to date are unknown. In a recent GWAS, Ferreira et al. identified a new association between asthma risk and common variants located on chromosome 8q21. The overarching aim of this thesis was to elucidate the biological mechanisms underlying this association. Specifically, the goals of this study were to identify the gene(s) underlying the observed association and to study their contribution to asthma pathophysiology. Using genetic data from the 1000 Genomes Project, we first identified 118 variants in linkage disequilibrium (LD; r2>0.6) with the sentinel allergy risk SNP (rs7009110) on chromosome 8q21. Of these, 35 were found to overlap one of four Putative Regulatory Elements (PREs) identified in this region in a lymphoblastoid cell line (LCL), based on epigenetic marks measured by the ENCODE project. Results from analysis of gene expression data generated for LCLs (n=373) by the Geuvadis consortium indicated that rs7009110 is associated with the expression of only one nearby gene: PAG1 - located 732 kb away. PAG1 encodes a transmembrane adaptor protein localized to lipid rafts, which is highly expressed in immune cells. Results from chromosome conformation capture (3C) experiments showed that PREs in the region of association physically interacted with the promoter of PAG1. -

Predicting Gene Ontology Biological Process from Temporal Gene Expression Patterns Astrid Lægreid,1,4 Torgeir R
Methods Predicting Gene Ontology Biological Process From Temporal Gene Expression Patterns Astrid Lægreid,1,4 Torgeir R. Hvidsten,2 Herman Midelfart,2 Jan Komorowski,2,3,4 and Arne K. Sandvik1 1Department of Cancer Research and Molecular Medicine, Norwegian University of Science and Technology, N-7489 Trondheim, Norway; 2Department of Information and Computer Science, Norwegian University of Science and Technology, N-7491 Trondheim, Norway; 3The Linnaeus Centre for Bioinformatics, Uppsala University, SE-751 24 Uppsala, Sweden The aim of the present study was to generate hypotheses on the involvement of uncharacterized genes in biological processes. To this end,supervised learning was used to analyz e microarray-derived time-series gene expression data. Our method was objectively evaluated on known genes using cross-validation and provided high-precision Gene Ontology biological process classifications for 211 of the 213 uncharacterized genes in the data set used. In addition,new roles in biological process were hypothesi zed for known genes. Our method uses biological knowledge expressed by Gene Ontology and generates a rule model associating this knowledge with minimal characteristic features of temporal gene expression profiles. This model allows learning and classification of multiple biological process roles for each gene and can predict participation of genes in a biological process even though the genes of this class exhibit a wide variety of gene expression profiles including inverse coregulation. A considerable number of the hypothesized new roles for known genes were confirmed by literature search. In addition,many biological process roles hypothesi zed for uncharacterized genes were found to agree with assumptions based on homology information. -

Stranded DNA and Sensitizes Human Kidney Renal Clear Cell Carcinoma
RESEARCH ARTICLE Exosome component 1 cleaves single- stranded DNA and sensitizes human kidney renal clear cell carcinoma cells to poly(ADP-ribose) polymerase inhibitor Qiaoling Liu1†, Qi Xiao1†, Zhen Sun1†, Bo Wang2†, Lina Wang1, Na Wang1, Kai Wang1, Chengli Song1*, Qingkai Yang1* 1Institute of Cancer Stem Cell, DaLian Medical University, Dalian, China; 2Department of General Surgery, Second Affiliated Hospital, DaLian Medical University, Dalian, China Abstract Targeting DNA repair pathway offers an important therapeutic strategy for Homo sapiens (human) cancers. However, the failure of DNA repair inhibitors to markedly benefit patients necessitates the development of new strategies. Here, we show that exosome component 1 (EXOSC1) promotes DNA damages and sensitizes human kidney renal clear cell carcinoma (KIRC) cells to DNA repair inhibitor. Considering that endogenous source of mutation (ESM) constantly assaults genomic DNA and likely sensitizes human cancer cells to the inhibitor, we first analyzed the statistical relationship between the expression of individual genes and the mutations for KIRC. Among the candidates, EXOSC1 most notably promoted DNA damages and subsequent mutations via preferentially cleaving C site(s) in single-stranded DNA. Consistently, EXOSC1 was more *For correspondence: significantly correlated with C>A transversions in coding strands than these in template strands in [email protected] human KIRC. Notably, KIRC patients with high EXOSC1 showed a poor prognosis, and EXOSC1 (CS); sensitized human cancer cells to poly(ADP-ribose) polymerase inhibitors. These results show that [email protected] (QY) EXOSC1 acts as an ESM in KIRC, and targeting EXOSC1 might be a potential therapeutic strategy. †These authors contributed equally to this work Competing interests: The Introduction authors declare that no DNA damages and subsequent mutations are central to development, progression, and treatment competing interests exist.