Oncogenic KRAS and BRAF Drive Metabolic Reprogramming in Colorectal Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Laboratory Genetic Testing in Clinical Practice
BioMed Research International Laboratory Genetic Testing in Clinical Practice Guest Editors: Ozgur Cogulu, Yasemin Alanay, and Gokce A. Toruner Laboratory Genetic Testing in Clinical Practice BioMed Research International Laboratory Genetic Testing in Clinical Practice Guest Editors: Ozgur Cogulu, Yasemin Alanay, and Gokce A. Toruner Copyright © 2013 Hindawi Publishing Corporation. All rights reserved. This is a special issue published in “BioMed Research International.” All articles are open access articles distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Contents Laboratory Genetic Testing in Clinical Practice,OzgurCogulu,YaseminAlanay,andGokceA.Toruner Volume 2013, Article ID 532897, 1 page Identification and Characterization of DM1 Patients by a NewDiagnostic Certified Assay: Neuromuscular and Cardiac Assessments, Rea Valaperta, Valeria Sansone, Fortunata Lombardi, Chiara Verdelli, Alessio Colombo, Massimiliano Valisi, Elisa Brigonzi, Elena Costa, and Giovanni Meola Volume 2013, Article ID 958510, 6 pages Ultradeep Pyrosequencing of Hepatitis C Virus Hypervariable Region 1 in Quasispecies Analysis, Kamila Caraballo Cortes,´ Osvaldo Zagordi, Tomasz Laskus, Rafal Ploski, Iwona Bukowska-O´sko, Agnieszka Pawelczyk, Hanna Berak, and Marek Radkowski Volume 2013, Article ID 626083, 10 pages Feasibility of a Microarray-Based Point-of-Care CYP2C19 Genotyping Test for Predicting Clopidogrel On-Treatment Platelet -

Anti-IDH3A Antibody (ARG42205)
Product datasheet [email protected] ARG42205 Package: 100 μl anti-IDH3A antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes IDH3A Tested Reactivity Hu, Ms, Rat Tested Application ICC/IF, IHC-P, WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name IDH3A Antigen Species Human Immunogen Recombinant fusion protein corresponding to aa. 28-366 of Human IDH3A (NP_005521.1). Conjugation Un-conjugated Alternate Names Isocitrate dehydrogenase [NAD] subunit alpha, mitochondrial; EC 1.1.1.41; NAD; Isocitric dehydrogenase subunit alpha; + Application Instructions Application table Application Dilution ICC/IF 1:50 - 1:200 IHC-P 1:50 - 1:200 WB 1:500 - 1:2000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Positive Control Raji Calculated Mw 40 kDa Observed Size ~ 40 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.3), 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw www.arigobio.com 1/3 cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Gene Symbol IDH3A Gene Full Name isocitrate dehydrogenase 3 (NAD+) alpha Background Isocitrate dehydrogenases catalyze the oxidative decarboxylation of isocitrate to 2-oxoglutarate. -

An Introduction to DKFZ
An Introduction to DKFZ Otmar D. Wiestler Deutsches Krebsforschungszentrum, Heidelberg Page 1 DKFZ Locations on Campus Cancer Research @ DKFZ • Established in 1964 • National research center • Helmholtz association • Location on Heidelberg biomedical campus • Team of 3000 members • Funding from BMBF & BW • Coordination of national & international activities Helmholtz International Graduate School for Cancer Research • High quality, comprehensive, structured and interdisciplinary graduate training in cancer research • Vivid interaction with international partners (Karolinska, MDACC, WIS) • Cooperation with Universites and Helmholtz centers 450 graduate students National Reference Center for Cancer Information New and expanding research fields Cancer genome Epigenetics, Small regulatory RNAs Systems biology of cancer & modelling Angiogenesis Cancer stem cells Metabolic dysfunction & cancer Cancer immunotherapies Neurooncology High field MRI, molecular imaging Particle therapy, radiation biology Epidemiology, early detection & prevention Stem cells & Cancer • Cancers originate from somatic stem or progenitor cells • Cancer cells with stem cell properties control tumor growth and biology • Cancer stem cells serve as a reservoir for tumor progression and recurrence DKFZ Cancer Stem Cell Program Joint activity with the Dietmar-Hopp-Foundation Three HI-STEM Junior Research Groups Marieke Essers Michael Milsom Christoph Rösli Cancer Genome Consortium • High number of genomic alterations in cancer cells • Striking inter-individual variability -

Mouse Idh3a Mutations Cause Retinal Degeneration and Reduced Mitochondrial Function Amy S
© 2018. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2018) 11, dmm036426. doi:10.1242/dmm.036426 RESEARCH ARTICLE Mouse Idh3a mutations cause retinal degeneration and reduced mitochondrial function Amy S. Findlay1, Roderick N. Carter2, Becky Starbuck3, Lisa McKie1, Klára Nováková1, Peter S. Budd1, Margaret A. Keighren1, Joseph A. Marsh1, Sally H. Cross1, Michelle M. Simon3, Paul K. Potter3, Nicholas M. Morton2 and Ian J. Jackson1,4,* ABSTRACT these diseases have a wide range of pathologies (Koopman et al., Isocitrate dehydrogenase (IDH) is an enzyme required for the 2012). Common to many of the diseases, however, are neurological production of α-ketoglutarate from isocitrate. IDH3 generates the or neuromuscular manifestations, including retinal disease. We have NADH used in the mitochondria for ATP production, and is a tetramer identified a missense mutation in the gene encoding a subunit of the made up of two α, one β and one γ subunit. Loss-of-function and tricarboxylic acid (TCA) cycle enzyme isocitrate dehydrogenase 3 missense mutations in both IDH3A and IDH3B have previously been (IDH3) as causing a retinal degeneration phenotype in mice. There implicated in families exhibiting retinal degeneration. Using mouse are three isocitrate dehydrogenase isozymes in mammals. IDH1 and models, we investigated the role of IDH3 in retinal disease and IDH2 are both homodimers that catalyse the decarboxylation of α mitochondrial function. We identified mice with late-onset retinal isocitrate to -ketoglutarate with the concomitant reduction of NADP degeneration in a screen of ageing mice carrying an ENU-induced to NADPH. IDH1 is cytoplasmic, whereas IDH2 is localised to the mutation, E229K, in Idh3a. -

Role of MDH2 Pathogenic Variant in Pheochromocytoma and Paraganglioma Patients
ARTICLE © American College of Medical Genetics and Genomics Role of MDH2 pathogenic variant in pheochromocytoma and paraganglioma patients Bruna Calsina, MSc, Mercedes Robledo, PhD et al.# Purpose: MDH2 (malate dehydrogenase 2) has recently been variant (c.429+1G>T). All were germline and those with available proposed as a novel potential pheochromocytoma/paraganglioma biochemical data, corresponded to noradrenergic PPGL. (PPGL) susceptibility gene, but its role in the disease has not been MDH2 MDH2 Conclusion: This study suggests that pathogenic variants addressed. This study aimed to determine the prevalence of may play a role in PPGL susceptibility and that they might be pathogenic variants among PPGL patients and determine the responsible for less than 1% of PPGLs in patients without associated phenotype. pathogenic variants in other major PPGL driver genes, a prevalence Methods: Eight hundred thirty patients with PPGLs, negative for similar to the one recently described for other PPGL genes. the main PPGL driver genes, were included in the study. However, more epidemiological data are needed to recommend Interpretation of variants of unknown significance (VUS) was MDH2 testing in patients negative for other major PPGL genes. performed using an algorithm based on 20 computational predictions, by implementing cell-based enzymatic and immuno- Genetics in Medicine (2018) 20:1652–1662; https://doi.org/10.1038/ fluorescence assays, and/or by using a molecular dynamics s41436-018-0068-7 simulation approach. Results: Five variants with potential -

In Silico Analysis of IDH3A Gene Revealed Novel Mutations Associated with Retinitis Pigmentosa
bioRxiv preprint doi: https://doi.org/10.1101/554196; this version posted February 18, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. In silico analysis of IDH3A gene revealed Novel mutations associated with Retinitis Pigmentosa Thwayba A. Mahmoud1*, Abdelrahman H. Abdelmoneim1, Naseem S. Murshed1, Zainab O. Mohammed2, Dina T. Ahmed1, Fatima A. Altyeb1, Nuha A. Mahmoud3, Mayada A. Mohammed1, Fatima A. Arayah1, Wafaa I. Mohammed1, Omnia S. Abayazed1, Amna S. Akasha1, Mujahed I. Mustafa1,4, Mohamed A. Hassan1 1- Department of Biotechnology, Africa city of Technology, Sudan 2- Hematology Department, Ribat University Hospital, Sudan 3- Biochemistry Department, faculty of Medicine, National University, Sudan 4- Department of Biochemistry, University of Bahri, Sudan *Corresponding Author: Thwayba A. Mahmoud, Email: [email protected] Abstract: Background: Retinitis Pigmentosa (RP) refers to a group of inherited disorders characterized by the death of photoreceptor cells leading to blindness. The aim of this study is to identify the pathogenic SNPs in the IDH3A gene and their effect on the structure and function of the protein. Method: we used different bioinformatics tools to predict the effect of each SNP on the structure and function of the protein. Result: 20 deleterious SNPs out of 178 were found to have a damaging effect on the protein structure and function. Conclusion: this is the first in silico analysis of IDH3A gene and 20 novel mutations were found using different bioinformatics tools, and they could be used as diagnostic markers for Retinitis Pigmentosa. -

Views for Entrez
BASIC RESEARCH www.jasn.org Phosphoproteomic Analysis Reveals Regulatory Mechanisms at the Kidney Filtration Barrier †‡ †| Markus M. Rinschen,* Xiongwu Wu,§ Tim König, Trairak Pisitkun,¶ Henning Hagmann,* † † † Caroline Pahmeyer,* Tobias Lamkemeyer, Priyanka Kohli,* Nicole Schnell, †‡ †† ‡‡ Bernhard Schermer,* Stuart Dryer,** Bernard R. Brooks,§ Pedro Beltrao, †‡ Marcus Krueger,§§ Paul T. Brinkkoetter,* and Thomas Benzing* *Department of Internal Medicine II, Center for Molecular Medicine, †Cologne Excellence Cluster on Cellular Stress | Responses in Aging-Associated Diseases, ‡Systems Biology of Ageing Cologne, Institute for Genetics, University of Cologne, Cologne, Germany; §Laboratory of Computational Biology, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, Maryland; ¶Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand; **Department of Biology and Biochemistry, University of Houston, Houston, Texas; ††Division of Nephrology, Baylor College of Medicine, Houston, Texas; ‡‡European Molecular Biology Laboratory–European Bioinformatics Institute, Hinxton, Cambridge, United Kingdom; and §§Max Planck Institute for Heart and Lung Research, Bad Nauheim, Germany ABSTRACT Diseases of the kidney filtration barrier are a leading cause of ESRD. Most disorders affect the podocytes, polarized cells with a limited capacity for self-renewal that require tightly controlled signaling to maintain their integrity, viability, and function. Here, we provide an atlas of in vivo phosphorylated, glomerulus- expressed -
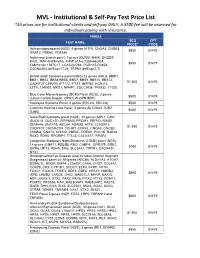
MVL - Institutional & Self-Pay Test Price List *All Prices Are for Institutional Clients and Self-Pay ONLY
MVL - Institutional & Self-Pay Test Price List *All prices are for institutional clients and self-pay ONLY. A $500 fee will be assessed for individuals paying with insurance. PANELS SEQ CPT TEST NAME PRICE* CODE Achromatopsiapanel (NGS), 6 genes (ATF6, CNGA3, CNGB3, $950 81479 GNAT2, PDE6C, PDE6H) Ashkenazi Jewish panel, 7 genes (CLRN1-N48K, DHDDS- K42E, MAK-K429insAlu, FAM161A-c.1355-6delCA, $550 81407 FAM161Ac.1567C>T, LCA5-Q279X, PCDH15-R245X, CACNA2D4-delExon17-26, TRPM1-delExon2-7) Bardet-Biedl Syndrome panel (NGS) 23 genes (ARL6, BBIP1, BBS1, BBS2, BBS4,BBS5, BBS7, BBS9, BBS10, BBS12, $1,500 81479 C8ORF37,CEP290, IFT172, IFT27, INPP5E, KCNJ13, LZTFL1,MKKS, MKS1, NPHP1, SDCCAG8, TRIM32, TTC8) Blue Cone Monochromacy (BCM) Panel (NGS), 2 genes $500 81479 (Locus Control Region, OPN1LW-OPN1MW) Cockayne Sydrome Panel, 2 genes (ERCC6, ERCC8) $500 81479 Common Hearing Loss Panel, 3 genes (SLC26A4, GJB2, $650 81479 GJB6) Cone-Rod Dystrophy panel (NGS), 33 genes (AIPL1, CRX, GUCA1A, GUCY2D, PITPNM3, PROM1, PRPH2, RIMS1, SEMA4A, UNC119, ABCA4, ADAM9, ATF6, C21ORF2, $1,950 81479 C8ORF37,CACNA2D4, CDHR1, CERKL, CNGA3, CNGB3, CNNM4, GNAT2, KCNV2, PDE6C, PDE6H, POC1B, RAB28, RAX2, RDH5, RPGRIP1, TTLL5, CACNA1F, RPGR) Congenital Stationary Night Blindness (CSNB) panel (NGS), 14 genes (GNAT1, PDE6B, RHO, CABP4, GPR179, GRK1, $950 81479 GRM6,LRIT3, RDH5, SAG, SLC24A1, TRPM1, CACNA1F, NYX) Developmental Eye Disease (also includes Anterior Segment Dysgenesis) panel v4: 59 genes (ABCB6, ALDH1A3, ATOH7, B3GALTL, BCOR, BMP4, c12orf57, CASK, CHD7, COL4A1, -

Gene Expression Studies of Hepatitis Virus-Induced Woodchuck Hepatocellular Carcinoma in Correlation with Human Results
33-44 6/12/06 18:31 Page 33 INTERNATIONAL JOURNAL OF ONCOLOGY 30: 33-44, 2007 33 Gene expression studies of hepatitis virus-induced woodchuck hepatocellular carcinoma in correlation with human results FANGJING WANG1, PAUL W. ANDERSON1, NICOLAS SALEM1, YU KUANG1, BUD C. TENNANT2 and ZHENGHONG LEE1,3 1Department of Biomedical Engineering, Case Western Reserve University, Cleveland, OH 44106; 2Department of Clinical Sciences, Cornell University, Ithaca, NY 14853; 3Department of Radiology, University Hospitals of Cleveland, Cleveland, OH 44106, USA Received July 19, 2006; Accepted September 20, 2006 Abstract. The lack of good molecular markers for diagnosis possible molecular imaging targets or biological markers in as well as treatment assessment has rendered the hepatocellular human HCC. carcinoma (HCC) a major challenge in health care. In this study, woodchucks were used as an animal model for hepatitis Introduction virus-induced HCC, and gene expression studies were per- formed using a human oligonucleotide microarray. An analysis Primary hepatocellular carcinoma (HCC) is the fifth most approach combing supervised significant analysis of microarray common malignancy in the world and the third most common (SAM), prediction analysis of microarray (PAM), and un- cause of cancer-related death (1). It is most prevalent in some supervised hierarchical cluster methodologies statistically of the Asian and African countries, but the incidence of determined 211 upregulated and 78 downregulated genes primary liver carcinoma in Western countries is on the rise between liver cancer and non-cancer liver tissues, and demon- in the past decades. HCC usually results from the chronic strated ≥93% accuracy in classifying the tissue samples. RT- inflammation caused by hepatitis B virus (HBV), hepatitis C PCR results confirmed the differential expression of selected virus (HCV), or long-term exposure to alcohol or dietary sequenced woodchuck genes (SAT, IDH3B, SCD) in the aflatoxin B1. -

Loss of the E3 Ubiquitin Ligase MKRN1 Represses Diet-Induced Metabolic Syndrome Through AMPK Activation
ARTICLE DOI: 10.1038/s41467-018-05721-4 OPEN Loss of the E3 ubiquitin ligase MKRN1 represses diet-induced metabolic syndrome through AMPK activation Min-Sik Lee1, Hyun-Ji Han2, Su Yeon Han2, Il Young Kim3,4, Sehyun Chae5, Choong-Sil Lee2, Sung Eun Kim2, Seul Gi Yoon4, Jun-Won Park4, Jung-Hoon Kim2, Soyeon Shin2, Manhyung Jeong2, Aram Ko2, Ho-Young Lee6, Kyoung-Jin Oh 7, Yun-Hee Lee 8, Kwang-Hee Bae7, Seung-Hoi Koo9, Jea-woo Kim10, Je Kyung Seong3,4, Daehee Hwang5 & Jaewhan Song 2 1234567890():,; AMP-activated protein kinase (AMPK) plays a key role in controlling energy metabolism in response to physiological and nutritional status. Although AMPK activation has been pro- posed as a promising molecular target for treating obesity and its related comorbidities, the use of pharmacological AMPK activators has been met with contradictory therapeutic challenges. Here we show a regulatory mechanism for AMPK through its ubiquitination and degradation by the E3 ubiquitin ligase makorin ring finger protein 1 (MKRN1). MKRN1 depletion promotes glucose consumption and suppresses lipid accumulation due to AMPK stabilisation and activation. Accordingly, MKRN1-null mice show chronic AMPK activation in both liver and adipose tissue, resulting in significant suppression of diet-induced metabolic syndrome. We demonstrate also its therapeutic effect by administering shRNA targeting MKRN1 into obese mice that reverses non-alcoholic fatty liver disease. We suggest that ubiquitin-dependent AMPK degradation represents a target therapeutic strategy for meta- bolic disorders. 1 Harvard Medical School, Boston Children’s Hospital, 3 Blackfan Circle CLS-16060.2, Boston, MA 02115, USA. 2 Department of Biochemistry, College of Life Science and Biotechnology, Yonsei University, Seoul 03722, Republic of Korea. -

Proteomic Survey Reveals Altered Energetic Patterns and Metabolic Failure Prior to Retinal Degeneration
The Journal of Neuroscience, February 19, 2014 • 34(8):2797–2812 • 2797 Neurobiology of Disease Proteomic Survey Reveals Altered Energetic Patterns and Metabolic Failure Prior to Retinal Degeneration Ana Griciuc,1 Michel J. Roux,2 Juliane Merl,1 Angela Giangrande,3 Stefanie M. Hauck,1 Liviu Aron,4 and Marius Ueffing1,5 1Research Unit Protein Science, Helmholtz Zentrum Mu¨nchen (GmbH)–German Research Center for Environmental Health, D-85764 Neuherberg, Germany, 2Department of Translational Medicine and Neurogenetics and 3Department of Development and Stem Cells, Institut de Ge´ne´tique et de Biologie Mole´culaire et Cellulaire, CNRS UMR 7104–INSERM U964–Universite´ de Strasbourg, 67404 Illkirch, France, 4Department of Genetics, Harvard Medical School, Boston, Massachusetts 02115, and 5Center for Ophthalmology, Institute for Ophthalmic Research, University of Tu¨bingen, 72076 Tu¨bingen, Germany Inherited mutations that lead to misfolding of the visual pigment rhodopsin (Rho) are a prominent cause of photoreceptor neuron (PN) degeneration and blindness. How Rho proteotoxic stress progressively impairs PN viability remains unknown. To identify the pathways that mediateRhotoxicityinPNs,weperformedacomprehensiveproteomicprofilingofretinasfromDrosophilatransgenicsexpressingRh1 P37H,the equivalentofmammalianRho P23H,themostcommonRhomutationlinkedtoblindnessinhumans.ProfilingofyoungRh1 P37H retinasrevealed acoordinatedupregulationofenergy-producingpathwaysandattenuationofenergy-consumingpathwaysinvolvingtargetofrapamycin(TOR) signaling,whichwasreversedinolderretinasattheonsetofPNdegeneration.WeprobedtherelevanceofthesemetabolicchangestoPNsurvival