Genome-Wide Analyses of Tandem Repeats and Transposable Elements in Patchouli
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Patchouli Essential Oil Extracted from Pogostemon Cablin (Blanco) Benth
Advances in Environmental Biology, 8(7) May 2014, Pages: 2301-2309 AENSI Journals Advances in Environmental Biology ISSN-1995-0756 EISSN-1998-1066 Journal home page: http://www.aensiweb.com/aeb.html Characterization and Antimicrobial Activity of Patchouli Essential Oil Extracted From Pogostemon cablin [Blanco] Benth. [lamiaceae] Ahmad Karimi Ph.D. in pharmacy, University of Santo Tomas, Philippines ARTICLE INFO ABSTRACT Article history: The physico-chemical properties of Philippine patchouli oil, hydro-distilled from fresh Received 25 March 2014 leaves and young shoots of Pogostemon cablin were characterized and found to be Received in revised form 20 April within the specifications set by the United States Essential Oils Society. Philippine 2014 patchouli oil and commercial patchouli oil have the same major components as shown Accepted 15 May 2014 by GC-MS analyses: patchouli alcohol, d-guaiene, a-guaiene, a-patchoulene, Available online 10 June 2014 seychellene, [3-patchoulene, and transcaryophylene, with slightly lower concentrations in the Philippine oil. Using the disk diffusion method patchouli oil was found to be Key words: active against the gram-positive bacteria: Staphylococcus, Bacillus, and Streptococcus Pogostemon cablin, patchouli oil, species. Fifty five percent [11/20] of community and only 14.8% [9/61] of hospital- essential oil, antimierobial activity, Staphylococcus aureus isolates were susceptible to an MIC of 0.03% [v/v.] and Sixty- physico-chemical properties four percent or 23/36 of methicillin-resistant Staphylococcus aureus [MRSA] isolates was sensitive to patchouli oil at 0.06%, as opposed to only 44% or 11/25 of the sensitive strains. Philippine patchouli essential oil was also active against several dermatophytes at 0.25%. -

Well-Known Plants in Each Angiosperm Order
Well-known plants in each angiosperm order This list is generally from least evolved (most ancient) to most evolved (most modern). (I’m not sure if this applies for Eudicots; I’m listing them in the same order as APG II.) The first few plants are mostly primitive pond and aquarium plants. Next is Illicium (anise tree) from Austrobaileyales, then the magnoliids (Canellales thru Piperales), then monocots (Acorales through Zingiberales), and finally eudicots (Buxales through Dipsacales). The plants before the eudicots in this list are considered basal angiosperms. This list focuses only on angiosperms and does not look at earlier plants such as mosses, ferns, and conifers. Basal angiosperms – mostly aquatic plants Unplaced in order, placed in Amborellaceae family • Amborella trichopoda – one of the most ancient flowering plants Unplaced in order, placed in Nymphaeaceae family • Water lily • Cabomba (fanwort) • Brasenia (watershield) Ceratophyllales • Hornwort Austrobaileyales • Illicium (anise tree, star anise) Basal angiosperms - magnoliids Canellales • Drimys (winter's bark) • Tasmanian pepper Laurales • Bay laurel • Cinnamon • Avocado • Sassafras • Camphor tree • Calycanthus (sweetshrub, spicebush) • Lindera (spicebush, Benjamin bush) Magnoliales • Custard-apple • Pawpaw • guanábana (soursop) • Sugar-apple or sweetsop • Cherimoya • Magnolia • Tuliptree • Michelia • Nutmeg • Clove Piperales • Black pepper • Kava • Lizard’s tail • Aristolochia (birthwort, pipevine, Dutchman's pipe) • Asarum (wild ginger) Basal angiosperms - monocots Acorales -

Species ANALYSIS International Journal for Species ISSN 2319 – 5746 EISSN 2319 – 5754
Species ANALYSIS International Journal for Species ISSN 2319 – 5746 EISSN 2319 – 5754 Diversity and therapeutic potentiality of the family Lamiaceae in Karnataka State, India: An overview Rama Rao V1҉, Shiddamallayya N2, Kavya N3, Kavya B4, Venkateshwarlu G5 1. Research Officer (Botany), Survey of Medicinal Plants Unit, National Ayurveda Dietetics Research Institute (CCRAS), Govt. Central Pharmacy Annexe, Ashoka Pillar, Jayangar, Bangalore-560011, India. 2. Assistant Research Officer (Botany), Survey of Medicinal Plants Unit, National Ayurveda Dietetics Research Institute (CCRAS), Govt. Central Pharmacy Annexe, Ashoka Pillar, Jayangar, Bangalore-560011, India. 3. Senior Research Fellow (Ayurveda), National Ayurveda Dietetics Research Institute (CCRAS), Govt. Central Pharmacy Annexe, Ashoka Pillar, Jayangar, Bangalore-560011, India. 4. Junior Research Fellow (Botany), National Ayurveda Dietetics Research Institute (CCRAS), Govt. Central Pharmacy Annexe, Ashoka Pillar, Jayangar, Bangalore-560011, India. 5. Research Officer (Scientist-3) in-charge, National Ayurveda Dietetics Research Institute (CCRAS), Govt. Central Pharmacy Annexe, Ashoka Pillar, Jayangar, Bangalore-560011, India. ҉Corresponding author: Survey of Medicinal Plants Unit, National Ayurveda Dietetics Research Institute (CCRAS), Govt. Central Pharmacy Annexe, Ashoka Pillar, Jayangar, Bangalore-560011, India, e-mail: [email protected] Publication History Received: 25 November 2014 Accepted: 11 January 2015 Published: 4 March 2015 Citation Rama Rao V, Shiddamallayya N, Kavya N, Kavya B, Venkateshwarlu G. Diversity and therapeutic potentiality of the family Lamiaceae in Karnataka State, India: An overview. Species, 2015, 13(37), 6-14 ABSTRACT The objective of the present study was to review the potential medicinal plants of Lamiaceae distributed throughout the state of Karnataka, India. Lamiaceae, also called as mint family is one of the largest families including herbs or shrubs often with aroma. -

Lamiales Newsletter
LAMIALES NEWSLETTER LAMIALES Issue number 4 February 1996 ISSN 1358-2305 EDITORIAL CONTENTS R.M. Harley & A. Paton Editorial 1 Herbarium, Royal Botanic Gardens, Kew, Richmond, Surrey, TW9 3AE, UK The Lavender Bag 1 Welcome to the fourth Lamiales Universitaria, Coyoacan 04510, Newsletter. As usual, we still Mexico D.F. Mexico. Tel: Lamiaceae research in require articles for inclusion in the +5256224448. Fax: +525616 22 17. Hungary 1 next edition. If you would like to e-mail: [email protected] receive this or future Newsletters and T.P. Ramamoorthy, 412 Heart- Alien Salvia in Ethiopia 3 and are not already on our mailing wood Dr., Austin, TX 78745, USA. list, or wish to contribute an article, They are anxious to hear from any- Pollination ecology of please do not hesitate to contact us. one willing to help organise the con- Labiatae in Mediterranean 4 The editors’ e-mail addresses are: ference or who have ideas for sym- [email protected] or posium content. Studies on the genus Thymus 6 [email protected]. As reported in the last Newsletter the This edition of the Newsletter and Relationships of Subfamily Instituto de Quimica (UNAM, Mexi- the third edition (October 1994) will Pogostemonoideae 8 co City) have agreed to sponsor the shortly be available on the world Controversies over the next Lamiales conference. Due to wide web (http://www.rbgkew.org. Satureja complex 10 the current economic conditions in uk/science/lamiales). Mexico and to allow potential partici- This also gives a summary of what Obituary - Silvia Botta pants to plan ahead, it has been the Lamiales are and some of their de Miconi 11 decided to delay the conference until uses, details of Lamiales research at November 1998. -

Indigenous Uses of Wild and Tended Plant Biodiversity Maintain Ecosystem Services in Agricultural Landscapes of the Terai Plains of Nepal
Indigenous uses of wild and tended plant biodiversity maintain ecosystem services in agricultural landscapes of the Terai Plains of Nepal Jessica P. R. Thorn ( [email protected] ) University of York https://orcid.org/0000-0003-2108-2554 Thomas F. Thornton University of Oxford School of Geography and Environment Ariella Helfgott University of Oxford Katherine J. Willis University of Oxford Department of Zoology, University of Bergen Department of Biology, Kew Royal Botanical Gardens Research Keywords: agrobiodiversity conservation; ethnopharmacology; ethnobotany; ethnoecology; ethnomedicine; food security; indigenous knowledge; medicinal plants; traditional ecological knowledge Posted Date: April 16th, 2020 DOI: https://doi.org/10.21203/rs.2.18028/v3 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at Journal of Ethnobiology and Ethnomedicine on June 8th, 2020. See the published version at https://doi.org/10.1186/s13002-020-00382-4. Page 1/36 Abstract Background Despite a rapidly accumulating evidence base quantifying ecosystem services, the role of biodiversity in the maintenance of ecosystem services in shared human-nature environments is still understudied, as is how indigenous and agriculturally dependent communities perceive, use and manage biodiversity. The present study aims to document traditional ethnobotanical knowledge of the ecosystem service benets derived from wild and tended plants in rice- cultivated agroecosystems, compare this to botanical surveys, and analyse the extent to which ecosystem services contribute social-ecological resilience in the Terai Plains of Nepal. Method Sampling was carried out in four landscapes, 22 Village District Committees and 40 wards in the monsoon season. -

In Vitro Antibacterial Activity of 34 Plant Essential Oils Against Alternaria Alternata
E3S Web o f Conferences 136, 06006 (2019) https://doi.org/10.1051/e3sconf/20191360 6006 ICBTE 2019 In vitro antibacterial activity of 34 plant essential oils against Alternaria alternata Qiyu Lu1, Ji Liu2, Caihong Tu2, Juan Li3, Chunlong Lei3, Qiliang Guo2, Zhengzhou Zhang2, Wen Qin1* 1College of Food Science and Technology, Sichuan Agricultural University, Ya'an, Sichuan, 625014, China 2Institute of Food Science and Technology, Chengdu Academy of Agricultural and Forestry Science, Chengdu, Sichuan, 611130, China 3 Institute of Animal Science, Chengdu Academy of Agricultural and Forestry Science, Chengdu, Sichuan, 611130, China Abstract. To determine the antibacterial effect of 34 plant essential oils on Alternaria alternata, 34 plant essential oils such as asarum essential oil, garlic essential oil, and mustard essential oil are used as inhibition agents to isolate A. alternata from citrus as indicator bacteria, through the bacteriostasis test and drug susceptibility test, the types of essential oils with the best inhibitory effect were screened and their concentration was determined. The results showed that the best inhibition effect was mustard essential oil with a minimum inhibitory concentration of 250 μl/L and a minimum bactericidal concentration of 250 μl/L. Followed by the Litsea cubeba essential oil and basil oil, the minimum inhibitory concentration is 500 μl/L. 1 Introduction quality and safety. A. alternata is the main fungus causing post-harvest disease of Pitaya, and the Citrus is a general term for orange, mandarin, orange, antibacterial effect of using cinnamon oil and clove oil is kumquat, pomelo, and medlar. Citrus is the world's good [10]. Clove oil can effectively inhibit the growth of largest fruit, rich in sugar, organic acids, mineral A. -

Herbs, Spices and Essential Oils
Printed in Austria V.05-91153—March 2006—300 Herbs, spices and essential oils Post-harvest operations in developing countries UNITED NATIONS INDUSTRIAL DEVELOPMENT ORGANIZATION Vienna International Centre, P.O. Box 300, 1400 Vienna, Austria Telephone: (+43-1) 26026-0, Fax: (+43-1) 26926-69 UNITED NATIONS FOOD AND AGRICULTURE E-mail: [email protected], Internet: http://www.unido.org INDUSTRIAL DEVELOPMENT ORGANIZATION OF THE ORGANIZATION UNITED NATIONS © UNIDO and FAO 2005 — First published 2005 All rights reserved. Reproduction and dissemination of material in this information product for educational or other non-commercial purposes are authorized without any prior written permission from the copyright holders provided the source is fully acknowledged. Reproduction of material in this information product for resale or other commercial purposes is prohibited without written permission of the copyright holders. Applications for such permission should be addressed to: - the Director, Agro-Industries and Sectoral Support Branch, UNIDO, Vienna International Centre, P.O. Box 300, 1400 Vienna, Austria or by e-mail to [email protected] - the Chief, Publishing Management Service, Information Division, FAO, Viale delle Terme di Caracalla, 00100 Rome, Italy or by e-mail to [email protected] The designations employed and the presentation of material in this information product do not imply the expression of any opinion whatsoever on the part of the United Nations Industrial Development Organization or of the Food and Agriculture Organization of the United Nations concerning the legal or development status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. -

Development of Process for Purification of Α and Β-Vetivone from Vetiver Essential Oil & Investigation of Effects of Heavy
UNIVERSITY OF NEW SOUTH WALES PhD Thesis Proposal Development of process for purification of α and β-vetivone from Vetiver essential oil & Investigation of effects of heavy metals on quality and quantity of extracted Vetiver oil Thai Danh Luu {B. Agric. Sc. (Qld University, Australia), M. Sc. (VUB, Belgium)} School of Chemical Sciences and Engineering Unversity of New South Wales, Sydney 1 Supervisor: Prof Neil Foster Supervisor: Prof Neil Foster Co-supervisors: Prof Tam Tran 2 and Dr Paul Truong 3 1 School of Chemical Sciences and Engineering, UNSW 2 Division of Engineering, Science and Technology, UNSW-Asia 3 Veticon Consulting and International Vetiver Network, Brisbane 1 I. INTRODUCTION In recent years, there is an increasing trend in research of essential oils extracted from various herbs and aromatic plants due to the continuous discoveries of their multifunctional properties other than their classical roles as food additives and/or fragrances. New properties of many essential oils, such as antibacterial, antifungal, antioxidant, and anti-inflammatory activities have been found and confirmed (Aruoma et al, 1996; Hammer et al, 1999; Güllüce et al, 2003). The pharmacological properties of essential oils extracted from plants have received the greatest interest of academic institutes and pharmaceutical companies (Ryman, 1992; Loza- Tavera, 1999; Courreges et al, 2000; Carnesecchi et al, 2002 and Salim et al, 2003). On the other hand, the insecticidal activities of essential oils are more favored by agricultural scientists and agri-business. Consequently, many investigation and new findings have significantly prompted and expanded novel applications of essential oils which are now been widely used as natural insecticides, cosmeceuticals, and aroma therapeutic agents. -
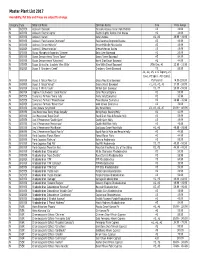
Master Plant List 2017.Xlsx
Master Plant List 2017 Availability, Pot Size and Prices are subject to change. Category Type Botanical Name Common Name Size Price Range N BREVER Azalea X 'Cascade' Cascade Azalea (Glenn Dale Hybrid) #3 49.99 N BREVER Azalea X 'Electric Lights' Electric Lights Double Pink Azalea #2 44.99 N BREVER Azalea X 'Karen' Karen Azalea #2, #3 39.99 - 49.99 N BREVER Azalea X 'Poukhanense Improved' Poukhanense Improved Azalea #3 49.99 N BREVER Azalea X 'Renee Michelle' Renee Michelle Pink Azalea #3 49.99 N BREVER Azalea X 'Stewartstonian' Stewartstonian Azalea #3 49.99 N BREVER Buxus Microphylla Japonica "Gregem' Baby Gem Boxwood #2 29.99 N BREVER Buxus Sempervirens 'Green Tower' Green Tower Boxwood #5 64.99 N BREVER Buxus Sempervirens 'Katerberg' North Star Dwarf Boxwood #2 44.99 N BREVER Buxus Sinica Var. Insularis 'Wee Willie' Wee Willie Dwarf Boxwood Little One, #1 13.99 - 21.99 N BREVER Buxus X 'Cranberry Creek' Cranberry Creek Boxwood #3 89.99 #1, #2, #5, #15 Topiary, #5 Cone, #5 Spiral, #10 Spiral, N BREVER Buxus X 'Green Mountain' Green Mountain Boxwood #5 Pyramid 14.99-299.99 N BREVER Buxus X 'Green Velvet' Green Velvet Boxwood #1, #2, #3, #5 17.99 - 59.99 N BREVER Buxus X 'Winter Gem' Winter Gem Boxwood #5, #7 59.99 - 99.99 N BREVER Daphne X Burkwoodii 'Carol Mackie' Carol Mackie Daphne #2 59.99 N BREVER Euonymus Fortunei 'Ivory Jade' Ivory Jade Euonymus #2 35.99 N BREVER Euonymus Fortunei 'Moonshadow' Moonshadow Euonymus #2 29.99 - 35.99 N BREVER Euonymus Fortunei 'Rosemrtwo' Gold Splash Euonymus #2 39.99 N BREVER Ilex Crenata 'Sky Pencil' -

Molecular Characterization of Patchouli (Pogostemon Spp) Germplasm
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Repositório Institucional da Universidade Federal de Sergipe Molecular characterization of patchouli (Pogostemon spp) germplasm S.S. Sandes1, M.I. Zucchi2, J.B. Pinheiro3, M.M. Bajay3, C.E.A. Batista3, F.A. Brito1, M.F. Arrigoni-Blank1, S.V. Alvares-Carvalho1, R. Silva-Mann1 and A.F. Blank1 1Laboratório de Recursos Genéticos e Óleos Essenciais, Departamento de Engenharia Agronômica, Universidade Federal de Sergipe, São Cristóvão, SE, Brasil 2Laboratório de Biologia Molecular, Agência Paulista de Tecnologia dos Agronegócios, Polo Centro Sul, Piracicaba, SP, Brasil 3Laboratório de Diversidade Genética e Melhoramento, Departamento de Genética, Escola Superior de Agricultura “Luiz de Queiroz”, Universidade de São Paulo, Piracicaba, SP, Brasil Corresponding author: A.F. Blank E-mail: [email protected] Genet. Mol. Res. 15 (1): gmr.15017458 Received August 17, 2015 Accepted October 29, 2015 Published February 19, 2016 DOI http://dx.doi.org/10.4238/gmr.15017458 ABSTRACT. Patchouli [Pogostemon cablin (Blanco) Benth.] is an aromatic, herbaceous plant belonging to the Lamiaceae family native to Southeast Asia. Its leaves produce an essential oil regularly used by the perfume and cosmetics industries. However, since patchouli from the Philippines and India were described and named Pogostemon patchouli, there has been a divergence in the identity of these species. The objective of the current study was to study the genetic diversity of patchouli accessions in the Active Germplasm Bank of Universidade Federal de Sergipe using microsatellite and inter simple sequence repeat markers. The results of both types of molecular markers showed that there are two well-defined clusters of accessions that harbor exclusive alleles. -

Antimicrobial Activity of the Essential Oils of Three Species of Pogostemon
Journal Home page : www.jeb.co.in « E-mail : [email protected] JEB ISSN: 0254-8704 Journal of Environmental Biology CODEN: JEBIDP Antimicrobial activity of the essential oils of three species of Pogostemon J.E. Thoppil11 *, A. Tajo , J. Minija 1 , M.J. Deena 11 , K. Sreeranjini , L. Leeja 1 , M. Sivadasan 2 and A.H. Alfarhan 2 1Cell and Molecular Biology Division, Department of Botany, University of Calicut, Calicut–673 635, India 2Department of Botany and Microbiology, College of Science, King Saud University, P.B. No. 2455, Riyadh–11451, Kingdom of Saudi Arabia *Corresponding Authors Email : [email protected] Abstract Publication Info Antimicrobial studies on 7 strains of bacteria and 8 strains of fungi using disk diffusion assay, revealed Paper received: potential activities of crude essential oils in Pogostemon benghalensis, P. purpurascens and P. vestitus. 10 January 2013 Essential oils produced highest inhibition zones against Staphylococcus aureus (39.33, 37.33 and 35.67 mm for P. benghalensis essential oil) and Candida albicans (34.33, 26.33 and 17.67 mm for P. purpurascens essential oil) among bacteria and fungi respectively, when compared with pure reference Revised received: -1 25 July 2013 standards (35 mm for Gentamycin sulphate (40 mg ml ) against S. aureus and 30.33 mm for Nystatin [50 IU] against C. albicans). Results also indicated the existence of potential antimicrobial activity of Pogostemon essential oils against other microorganisms viz., Proteus vulgaris, E. coli and Aspergillus Re-revised received: parasiticus. Leaf essential oils of P. purpurascens and P. benghalensis can be considered as a new source 05 September 2013 for developing local antifungal and antibacterial agents. -

A Taxonomic Study of Lamiaceae (Mint Family) in Rajpipla (Gujarat, India)
World Applied Sciences Journal 32 (5): 766-768, 2014 ISSN 1818-4952 © IDOSI Publications, 2014 DOI: 10.5829/idosi.wasj.2014.32.05.14478 A Taxonomic Study of Lamiaceae (Mint Family) in Rajpipla (Gujarat, India) 12Bhavin A. Suthar and Rajesh S. Patel 1Department of Botany, Shri J.J.T. University, Vidyanagari, Churu-Bishau Road, Jhunjhunu, Rajasthan-333001 2Biology Department, K.K. Shah Jarodwala Maninagar, Science College, Ahmedabad Gujarat, India Abstract: Lamiaceae is well known for its medicinal herbs. It is well represented in Rajpipla forest areas in Gujarat State, India. However, data or information is available on these plants are more than 35 years old. There is a need to be make update the information in terms of updated checklist, regarding the morphological and ecological data and their distribution ranges. Hence the present investigation was taken up to fulfill the knowledge gap. In present work 13 species belonging to 8 genera are recorded including 8 rare species. Key words: Lamiaceae Rajpipla forest Gujarat INTRODUCTION recorded by masters. Many additional species have been described from this area. Shah [2] in his Flora of Gujarat The Lamiaceae is a very large plant family occurring state recoded 38 species under 17 genera for this family. all over the world in a wide variety of habitats from alpine Before that 5 genera and 7 species were recorded in First regions through grassland, woodland and forests to arid Forest flora of Gujarat [3]. and coastal areas. Plants are botanically identified by their Erlier “Rajpipla” was a small state in the British India; family name, genus and species.