S1 Supplementary Information the Large Subunit of the Regulatory [Nife
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
4M4t Netz0s 4U
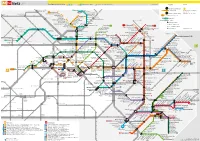
Netz Tarifbereich Berlin A B C A B Haltestellen in Berlin C Haltestellen in Brandenburg www.BVG.de Legende Service 4M 4t 0s 4u 24 Linie im 24-Stunden-Betrieb Waidmannslust :hE 50 M1 Guyotstr. Arnouxstr. S8 MetroTram täglich Berliner Verkehrsbetriebe (BVG) Wittenau :8 Navarraplatz M1 Hugenotten- S2 www.BVG.de Rosenthal Nord Französisch Buchholz Kirche platz 20 BVG Call Center: 030 19 44 9 Karl-Bonhoeffer- Hauptstr./Friedrich-Engels-Str. Linie im 20-Stunden-Betrieb Tegel Blankenfelder Str. Nervenklinik Wiesenwinkel Rosenthaler Str. M1 MetroTram-Abschnitte Schillerstr. M1 S25 Angerweg Marienstr./Pasewalker Str. Schönholz Waldemarstr. Nordendstr. Blankenburg 50 Straßenbahn Friedrich-Engels-Str./Eichenstr. Nordend Pasewalker Str./Blankenburger Weg Heinrich-Böll-Str. Wartenberg :gE 2 7 S+U-Bahn U6 Am Iderfenngraben Pankower Str. Kuckhoffstr. Umsteigemöglichkeit U8 Pastor-Niemöller-Platz H.-Hesse-Str./ . S1 S2 eg Galenusstr. Grabbeallee/Pastor-Niemöller-Platz Waldstr. > Halt nur in Pfeilrichtung 5 S8 Tschaikowskistr. S Pankow-Heinersdorf M4 M5 Zingster Str. 0A 128 StiftswMendelstr Heinersdorf M2 Falkenberg M4 M17 0b 5 Bürgerpark Pankow Fern- und Regionalbahnhof Kurt-Schumacher-Platz Würtzstr. Rothenbachstr. Zingster Str./Ribnitzer Str. Welsestr. :9U Osloer Str. Drontheimer Str. Rathaus Pankow Pankow Heinersdorf Kirche Falkenberger Ch./Prendener Str. Regionalbahnhof Stand: 3. April 2016 Kirche Ahrenshooper Str. Louise-Schroeder-Platz Am Wasserturm S Hohenschönhausen Osloer Str./ Prerower Platz Prinzenallee :9 :2 Tino-Schwierzina-Str. Osram-Höfe S+U Pankow Hansastr./Malchower Weg Rüdickenstr. 8 0 Am Steinberg M2 12 5 S2 S8 S9 M4 Arnimstr. :7 Ahrensfelde 3 M1 Masurenstr. Feldtmannstr. M1 3 Prenzlauer Promenade/ 5 Stadion Buschallee/Hansastr. 0 Grüntaler Str. Am Steinberg 12 27 Pasedagplatz U Seestr. -

Rail Transport in the World's Major Cities
Feature Evolution of Urban Railways (part 2) Rail Transport in The World’s Major Cities Takao Okamoto and Norihisa Tadakoshi Many of the world’s large cities grew in development, particularly with regard to American city; and Hong Kong, Seoul and conjunction with railways, and today, the following: Tokyo representing Asian cities. large cities cannot depend only on motor • The correlation between railways and vehicles for transportation. With worries urban growth over global environmental issues, public • The location of terminals for intercity London transportation systems are increasingly and intra-city transport seen as an important way to expand and • Examination of various public transit Located in southeast England near the revitalize large cities, while consuming systems, including non-infrastructure mouth of the River Thames, London less energy and other resources. • Strategic planning of rail networks expanded during the Industrial Revolution This article looks at public transportation based on urban development trends (1760–1850) and secured its dominance systems in some major cities of the world and future models as the heart of the British Empire during and identifies similarities and differences The cities selected for this comparative the Victorian era (1837–1901). It’s in areas such as history of development, study are: London as the first city to adapt population grew from about 500,000 in railway networks, and method of rail technology to public transport; Paris, the 17th century to 4.5 million by the end operation. Our aim is to explore the future Berlin and Moscow as three major of the 19th century. The modern relationship between urban and transport European cities; New York as a North- metropolis of Greater London consists of The London Railway Network King’s Cross/St.Pancras Paddington LCY Victoria Waterloo LHR R. -

Berlin Metro Map by Zuti
Hohen Mühlenbeck Bernau Borgsdorf Neuendorf Bergfelde Schönfließ Mönchmühle Karow Röntgental Friedenstal Oranienburg Bernau Lehnitz Birkenwerder Hugenotten Navarrapl Buch Zepernick Guyotstr bei Bernau Rosenthal Nord Arnoux HAVEL Französisch Hauptstr Buchholz Kirche Frohnau Friedrich Engels 50 HAVEL Wiesenwinkel Blankenfelder Berlin Angerweg © Copyright Visual IT Ltd Nordendstr Rosenthaler ® Zuti and the Zuti logo are registered trademarks Hermsdorf www.zuti.co.uk Nordend Schillerstr Marienstr BERLIN WALL BERLIN Uhlandstr Pasewalker Blankenburg Hennigsdorf Waldemar Waidmannslust Pasewalker Platanenstr Heinrich Böll Blankenburger Weg Heiligensee Pankower Am Iderfenngraben Kuckhoffstr Pastor Niemöller Platz Schulzendorf Galenusstr Wittenau Hermann Hesse Grabbeallee Waldstr Pastor Niemöller Ahrensfelde REINICKENDORF Ahrensfelde Tschaikowskistr HAVEL Rathaus Würtzstr Wartenberg Reinickendorf Mendelstr Tegel Wilhelmsruh M1 Pankow Zingster Falkenberger Karl B Heinersdorf Prendener Welsestr Nerven Bürgerpark Stiftsweg Heinersdorf Falkenberg Barnimplatz Alt Tegel klinik Alt Reinickendorf Pankow Rathaus Zingster Ribnitzer Schönholz Pankow PANKOW Hohenschönhausen Eichborn Ahrenshooper Niemegker Borsigwerke damm Pankow Rothenbachstr Paracelsus Bad Kirche Prerower U8 Mehrower Holzhauser Lindauer Hansastr Malchower Wuhletalstr HAVEL Heinersdorf Kirche Otisstr Allee Wollankstr JUNGFERNHEIDE Residenzstr Pankow Feldtmannstr Rüdickenstr Max Hermann TEGELER SEE Am Wasserturm M5 Scharnweber Masurenstr M2 Pasedagplatz Berliner Allee Franz Neumann Am Steinberg -

Train Simulator - Right Through Berlin
Train Simulator - Right through Berlin © Historical S-Bahn Berlin e.V. 2020 Page 1 Train Simulator - Right through Berlin Manual "Mitten durch Berlin" - Operation Series 476 Table of contents Table of contents ....................................................................................................... 2 0. authors ................................................................................................................ 3 1. introduction .......................................................................................................... 3 2. system requirements .............................................................................................. 4 3. the driver's cab ..................................................................................................... 5 4. upgrading .......................................................................................................... 10 4.1 Train is cold (full upgrade) .............................................................................. 10 4.2 Transfer of trains from other Tf ......................................................................... 10 4.3 Train is fully upgraded .................................................................................... 10 4.4 Change of driver's cab ................................................................................... 11 5. door control ....................................................................................................... 11 6. driving and braking ........................................................................................... -

Studie Zu ÖPNV Engpässen Und Lösungen
Studie zu aktuellen und prognosti- Auftraggeber: schen Engpässen und Lösungen im IHK Industrie- und Handelskammer Berliner ÖPNV Berlin Fasanenstraße 85 10623 Berlin https://www.ihk-berlin.de/ Bericht, April 2018 Auftragnehmer: VCDB VerkehrsConsult Dresden-Berlin GmbH Uhlandstraße 97 10715 Berlin Könneritzstraße 31 01067 Dresden Tel.: 0351 / 4 82 31 00 Fax: 0351 / 4 82 31 09 E-Mail: [email protected] Web: http://www.vcdb.de Deutsches Zentrum für Luft- und Raumfahrt e.V. (DLR) Institut für Verkehrsforschung Rutherfordstraße 2 12489 Berlin Tel.: 030 67055-681 Fax: 030 67055-283 Web: www.DLR.de/vf Ansprechpartner: Lutz Richter E-Mail: [email protected] Dr. Matthias Heinrichs E-Mail: [email protected] Studie zu aktuellen und prognostischen Engpässen und Lösungen im Berliner ÖPNV Inhaltsverzeichnis Inhaltsverzeichnis 1 Ausgangslage und Zielstellung ................................... 7 2 ÖPNV-Analyse ............................................................. 8 2.1 Aufbau des Verkehrsmodells ................................................ 8 2.2 Fahrzeuganalyse ................................................................... 9 2.3 Prognosenullfall 2030 .......................................................... 11 3 Prognose der Nachfrageentwicklung absehbarer Engpässe im Berliner ÖPNV ..................................... 13 3.1 Methodik .............................................................................. 13 3.2 Verkehrsumlegung 2030...................................................... 17 4 Empfehlungen zur Lösung der -

Adding Transit to an Agent-Based Transportation Simulation: Concepts and Implementation
Adding Transit to an Agent-Based Transportation Simulation Concepts and Implementation vorgelegt von Dipl. Inf.-Ing. ETH Marcel Rieser aus Affeltrangen (TG), Schweiz von der Fakultät V, Verkehrs- und Maschinensysteme der Technischen Universität Berlin zur Erlangung des akademischen Grades eines Doktor-Ingenieurs (Dr.-Ing.) genehmigte Dissertation Promotionsausschuss: Vorsitzende: Prof. Dr.-Ing. Chr. Ahrend Gutachter: Prof. Dr. rer. nat. Kai Nagel Gutachter: Prof. Dr.-Ing. Kay W. Axhausen Tag der wissenschaftlichen Aussprache: 10. Juni 2010 Berlin 2010 D 83 Contents Abstract ix Kurzfassung xi 1 Introduction 1 2 Public Transportation Systems 5 2.1 Definitions ............................... 5 2.2 Public Transportation in Developed Countries ................. 6 2.2.1 Overview ............................. 6 2.2.2 Data Requirements ........................ 7 2.3 Public Transportation in Developing Countries ................ 9 2.3.1 Overview ............................. 9 2.3.2 Data Requirements ........................ 9 3 Related Work 11 3.1 Transportation Simulation in General .................... 11 3.2 Multi-Modal and Transit Simulation ..................... 13 3.2.1 Transit Assignment ........................ 13 3.2.2 Multimodal Route Choice ...................... 14 3.2.3 Operational Simulation ....................... 15 3.2.4 Multi-Modal Simulation ....................... 16 4 Agent-Based Transportation Simulation 19 4.1 Overview ................................ 19 4.2 Controler ................................ 22 4.3 Initial Demand -

Transit Systems in the Us and Germany
TRANSIT SYSTEMS IN THE US AND GERMANY - A COMPARISON A Thesis Presented to The Academic Faculty by Johannes von dem Knesebeck In Partial Fulfillment of the Requirements for the Degree Master of Science in Civil Engineering in the School of Civil and Environmental Engineering Georgia Institute of Technology August 2011 TRANSIT SYSTEMS IN THE US AND GERMANY - A COMPARISON Approved by: Dr. Michael D. Meyer, Advisor School of Civil and Environmental Engineering Georgia Institute of Technology Dr. Adjo Akpene Amekudzi School of Civil and Environmental Engineering Georgia Institute of Technology Dr. Frank Southworth School of Civil and Environmental Engineering Georgia Institute of Technology Date Approved: July 5, 2011 ACKNOWLEDGEMENTS I wish to thank my advisor Dr. Michael D. Meyer for his help and constant support during the writing of this thesis. I also wish to thank the members of my thesis committee Dr. Adjo A. Amekudzi and Dr. Frank Southworth for their helpful comments and input. Furthermore, I would like to thank all the respective transit agencies in Germany and the US for making data available to me and helping me with hints and comments about my research. iii TABLE OF CONTENTS Page ACKNOWLEDGEMENTS iii LIST OF TABLES vii LIST OF FIGURES viii LIST OF SYMBOLS AND ABBREVIATIONS ix SUMMARY xi CHAPTER 1 Introduction and Methodology 12 1.1 Introduction 12 1.2 Methodology 13 1.2.1 Choice of Cities 13 1.2.2 Choice of Transit Systems 14 1.2.3 Collected Data 16 1.2.4 Definition of Rail System-Terms 19 1.2.5 Data Interpretation 21 1.3 Organization -

Accommodation Guide
BIMM BERLIN ACCOMMODATION GUIDE WHERE TO LIVE EUROPE’S PUBLIC TRANSPORT MOST CONNECTED TYPES OF LETTING MUSIC COLLEGE MANAGING FINANCES 2017/18 BIMM-INSTITUTE.DE CONTENTS WHERE SHOULD I BE LOOKING? 3 CONTACT THE ACCOMMODATION TEAM PUBLIC TRANSPORT 4 Accommodation advice is offered by PUBLIC TRANSPORT MAP 6 the Student Support Team. While we don’t provide housing for students directly, we’re HOUSE HUNTING 7 here to help and advise along the way. PRIVATE LETTINGS 8 If you need to send anything to the Student Support Team, please send it to: LETTING AGENCIES 10 BIMM Berlin Warschauer Straße 70a SEARCHING FOR A FLAT 11 10243 Berlin, Germany HOUSE VIEWINGS 12 If you find you have questions about your finances that aren’t answered in this guide, WHAT YOU NEED TO KNOW 13 then please contact the Student Support Team: Tel: 0800 664 7529 Email: [email protected] BIMM Berlin Accommodation Guide 2016/17 WHERE SHOULD I BE LOOKING? BIMM Berlin’s address is: that you’ll need to take into consideration Warschauer Str. 70A the cost of travelling, and the time it will take 10243 Berlin to get into BIMM Berlin, when making any accommodation decisions. The college’s nearest U-Bahn station is Warschauer Str on the U1, closest stop 5 min We recommend the following areas, some walk. The S-Bahn lines S3, S5, S7 and S75 from of which are within walking distance of Alexander Platz. BIMM Berlin or a short commute: The M10 and M13 trams stop very close to the • Friedrichshain college, which is also served by the 240 and • Kreuzberg N40 bus routes. -
Zukunftsstrategien Der S-Bahn Berlin Gmbh
Neue Direktverbindung mit Linie S9 vom Flughafen Schönefeld über die Stadtbahn nach Spandau . Die S9 wird über die neu erbaute Südringkurve und über die Stadtbahn (ohne Halt in Ostkreuz) zwischen Flughafen Schönefeld und Spandau verkehren . Regionalverkehrslinien RE 1, RE 2, RE 7 und RB 14 halten neu in Ostkreuz 1 S-Bahn Berlin | 17.11.2017 Die Linien der Stadtbahn werden neu geordnet – und verkehren nach Spandau, S5 nach Westkreuz Spandau – Erkner . übernimmt zusammen mit S9 Linienast nach Spandau . Erkner – Ostbahnhof alle 10 Minuten, weiter nach Spandau alle 20 Oranienburg Birkenwerder Minuten Bernau . im Nachtverkehr Ostkreuz – Erkner alle 30 Minuten . Neue Abfahrzeiten im gesamten Linienverlauf Hennigsdorf Tegel Wartenberg Westkreuz – Strausberg Nord Ahrensfelde 5 Schönholz Strausberg Nord 3 9 Gesundbr. S5 beginnt und endet neu in Westkreuz Hbf Spandau Zoo Ostkreuz . Westkreuz – Mahlsdorf/Hoppegarten alle 10 Minuten, weiter nach Strausberg Strausberg/Strausberg Nord alle 20 Minuten 5 Ostbf . im Nachtverkehr Ostbahnhof – Strausberg Nord Südkreuz . neue Abfahrzeiten im gesamten Linienverlauf (jeweils 5 Minuten früher Wannsee Spindlersfeld Erkner in beiden Richtungen) Adlershof Grünau 3 Teltow Stadt 9 Potsdam Hbf Flughafen Berlin Brandenburg Spandau – Flughafen Schönefeld Königs Wusterhausen Blankenfelde . neue Direktverbindung aus dem Berliner Südosten auf die Stadtbahn und weiter nach Spandau . in Baumschulenweg günstiger Übergang von und nach Königs Wusterhausen mit S46 . verkehrt alle 20 Minuten, im Nachtverkehr alle 30 Minuten . neue Abfahrzeiten im gesamten Linienverlauf 2 S-Bahn Berlin | 17.11.2017 verbindet unverändert Potsdam mit Berlin, kann baubedingt nur bis Ostkreuz verkehren Potsdam Hbf – Ahrensfelde . tagsüber 10-Minuten-Takt auf gesamtem Laufweg Oranienburg . Ausweitung des Angebots auf der Stadtbahn im Abendverkehr: neu Birkenwerder zwischen Westkreuz und Ahrensfelde und zusätzlich auch an Samstagen Bernau 10-Minuten-Takt bis ca. -

X Rqx Rw X 8 X
xrQ xrW Landsberger Allee <> x8 xiT Gesundbrunnen 8. 4. 2019 (Mo) – 20. 5. 2019 (Mo) 4 Uhr 1.30 Uhr Ersatzverkehr mit Bussen Replacement service by bus Kalender Einschränkungen April 2019 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 Mo Di Mi Do Fr Sa So Mo Di Mi Do Fr Sa So Mo Di Mi Do Fr Sa So :dA :dB :8 :hE Ersatzverkehr Landsberger Allee <> Gesundbrunnen mit Bussen :dA :dB :8 :hE kein Greifswalder Str. <> Schönhauser Allee S-Bahnverkehr :dA :dB kein Schönhauser Allee <> Gesundbrunnen S-Bahnverkehr :8 :hE kein Schönhauser Allee <> Bornholmer Str. S-Bahnverkehr :8 :hE :9 S-Bahnverkehr nur Baumschulenweg <> Treptower Park im 20-Minutentakt Einschränkungen Mai 2019 29 30 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 Mo Di Mi Do Fr Sa So Mo Di Mi Do Fr Sa So Mo Di Mi Do Fr Sa So :dA :dB :8 :hE Ersatzverkehr Landsberger Allee <> Gesundbrunnen mit Bussen :dA dB 8 hE kein Greifswalder Str. <> Schönhauser Allee S-Bahnverkehr :dA :dB kein Schönhauser Allee <> Gesundbrunnen S-Bahnverkehr :8 :hE kein Schönhauser Allee <> Bornholmer Str. S-Bahnverkehr :8 :hE :9 S-Bahnverkehr nur Baumschulenweg <> Treptower Park im 20-Minutentakt An den gesetzlichen Feiertagen (19.4. – Karfreitag, 22.4. – Ostermontag und 1.5. Maifeiertag) fahren die Züge wie sonntags. In der Nächten vor den Feiertagen wird durchgehender Nachtverkehr angeboten. On public holidays (19.4 - Good Friday, 22.4 - Easter Monday and 1.5 May Day), the trains run like Sundays. -

TS2020 – Class 481
TS2020 – Class 481 Typ Train Simulator 2020 - Addon Autor Benjamin Ebrecht Version 1.04 Datum 01.08.2020 Kontakt [email protected] Preface Dear Customer, thank you for your interest in our product “Die Baureihe 481”. With this Add-on you have purchased a very special realisation, as it is our very first to be based upon a vehicle model series made from scratch in the DTG Train Simulator. We have created a completely new, modular script- system to replicate the specific functions of the Berlin S-Bahn, following in an appropriately long time of development. The time was well spent: next to many very detailed characteristics like flickering lights and correctly calculated switching sparks, we also could implement entirely new systems such as an independent simulation of passengers for realistically packed trains and an extendable, fully functioning IBIS (Integrated-Board-Information-System) for the destination display and announcements. Another specialty is the fact that, in addition to the vehicle, we also managed to put a whole route into the package. Cheers to Jan Bleiß! He allowed us take the S25 part of his Add-on route “Berlin- Leipzig” and fit it to our needs for this package. Together with him, we look forward to see the fully finished S25 (to Henningsdorf); announced for 2020, with this route our vehicle will be able to fully play its potential… As usually, we have added some local colour to our scenarios and as a further addition the S-Bahn classes 475 (“Stadtbahner) and 480 (“Toaster”) are included in the package. Please note the extensive contents and complex vehicle functions of this manual. -

S-Bahn Linie S8 Fahrpläne & Netzkarten
S-Bahn Linie S8 Fahrpläne & Netzkarten Grünau ◄ ► Frankfurter Allee Im Website-Modus Anzeigen Die S-Bahn Linie S8 (Grünau ◄ ► Frankfurter Allee) hat 7 Routen (1) Birkenwerder: 00:09 - 23:49 (2) Zeuthen: 00:05 - 23:25 Verwende Moovit, um die nächste Station der S-Bahn Linie S8 zu ƒnden und, um zu erfahren wann die nächste S-Bahn Linie S8 kommt. Richtung: Birkenwerder S-Bahn Linie S8 Fahrpläne 16 Haltestellen Abfahrzeiten in Richtung Birkenwerder LINIENPLAN ANZEIGEN Montag 00:09 - 23:49 Dienstag 00:09 - 23:49 Grünau Adlergestell/ Wassersportallee 550, Berlin Mittwoch 00:09 - 23:49 Adlershof Donnerstag 00:09 - 23:49 Adlergestell 254-256, Berlin Freitag 00:09 - 23:49 Betriebsbahnhof Schöneweide Samstag 00:09 - 23:49 Adlergestell 103, Berlin Sonntag 00:09 - 23:49 S Schöneweide Bhf Baumschulenweg S Baumschulenweg, Berlin S-Bahn Linie S8 Info Plänterwald Richtung: Birkenwerder Stationen: 16 Treptower Park Fahrtdauer: 65 Min Bernhard-Langwaldt-Weg, Berlin Linien Informationen: Grünau, Adlershof, Betriebsbahnhof Schöneweide, S Schöneweide Bhf, Ostkreuz Bhf Baumschulenweg, Plänterwald, Treptower Park, Sonntagstraße 37, Berlin Ostkreuz Bhf, Frankfurter Allee, Storkower Str., Landsberger Allee, Greifswalder Str., Prenzlauer Frankfurter Allee Allee, Schönhauser Allee, Bornholmer Str., Pankow Rigaer Straße 55 B, Berlin Storkower Str. Langer Jammer, Berlin Landsberger Allee Landsberger Allee 117, Berlin Greifswalder Str. Greifswalder Straße 81, Berlin Prenzlauer Allee Kanzowstraße 15, Berlin Schönhauser Allee Schönhauser Allee 75, Berlin Bornholmer