'Assessment of Network Module Identification Across Complex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

Small Cell Ovarian Carcinoma: Genomic Stability and Responsiveness to Therapeutics
Gamwell et al. Orphanet Journal of Rare Diseases 2013, 8:33 http://www.ojrd.com/content/8/1/33 RESEARCH Open Access Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics Lisa F Gamwell1,2, Karen Gambaro3, Maria Merziotis2, Colleen Crane2, Suzanna L Arcand4, Valerie Bourada1,2, Christopher Davis2, Jeremy A Squire6, David G Huntsman7,8, Patricia N Tonin3,4,5 and Barbara C Vanderhyden1,2* Abstract Background: The biology of small cell ovarian carcinoma of the hypercalcemic type (SCCOHT), which is a rare and aggressive form of ovarian cancer, is poorly understood. Tumourigenicity, in vitro growth characteristics, genetic and genomic anomalies, and sensitivity to standard and novel chemotherapeutic treatments were investigated in the unique SCCOHT cell line, BIN-67, to provide further insight in the biology of this rare type of ovarian cancer. Method: The tumourigenic potential of BIN-67 cells was determined and the tumours formed in a xenograft model was compared to human SCCOHT. DNA sequencing, spectral karyotyping and high density SNP array analysis was performed. The sensitivity of the BIN-67 cells to standard chemotherapeutic agents and to vesicular stomatitis virus (VSV) and the JX-594 vaccinia virus was tested. Results: BIN-67 cells were capable of forming spheroids in hanging drop cultures. When xenografted into immunodeficient mice, BIN-67 cells developed into tumours that reflected the hypercalcemia and histology of human SCCOHT, notably intense expression of WT-1 and vimentin, and lack of expression of inhibin. Somatic mutations in TP53 and the most common activating mutations in KRAS and BRAF were not found in BIN-67 cells by DNA sequencing. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -
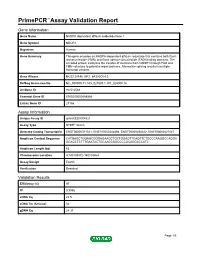
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name NADPH dependent diflavin oxidoreductase 1 Gene Symbol NDOR1 Organism Human Gene Summary This gene encodes an NADPH-dependent diflavin reductase that contains both flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) binding domains. The encoded protein catalyzes the transfer of electrons from NADPH through FAD and FMN cofactors to potential redox partners. Alternative splicing results in multiple transcript variants. Gene Aliases MGC138148, NR1, bA350O14.9 RefSeq Accession No. NC_000009.11, NG_027801.1, NT_024000.16 UniGene ID Hs.512564 Ensembl Gene ID ENSG00000188566 Entrez Gene ID 27158 Assay Information Unique Assay ID qHsaCED0004821 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000371521, ENST00000344894, ENST00000458322, ENST00000427047 Amplicon Context Sequence CATGAGCTGGAGCGGGAGAAGCTGCTGGAGTTCAGTTCTGCCCAAGGCCAGGA GGAGCTCTTTGAATACTGCAACCGGCCCCGCAGGACCATC Amplicon Length (bp) 63 Chromosome Location 9:140109372-140109464 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 97 R2 0.9996 cDNA Cq 22.5 cDNA Tm (Celsius) 82 gDNA Cq 24.37 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report NDOR1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 3/5 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Two Locus Inheritance of Non-Syndromic Midline Craniosynostosis Via Rare SMAD6 and 4 Common BMP2 Alleles 5 6 Andrew T
1 2 3 Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and 4 common BMP2 alleles 5 6 Andrew T. Timberlake1-3, Jungmin Choi1,2, Samir Zaidi1,2, Qiongshi Lu4, Carol Nelson- 7 Williams1,2, Eric D. Brooks3, Kaya Bilguvar1,5, Irina Tikhonova5, Shrikant Mane1,5, Jenny F. 8 Yang3, Rajendra Sawh-Martinez3, Sarah Persing3, Elizabeth G. Zellner3, Erin Loring1,2,5, Carolyn 9 Chuang3, Amy Galm6, Peter W. Hashim3, Derek M. Steinbacher3, Michael L. DiLuna7, Charles 10 C. Duncan7, Kevin A. Pelphrey8, Hongyu Zhao4, John A. Persing3, Richard P. Lifton1,2,5,9 11 12 1Department of Genetics, Yale University School of Medicine, New Haven, CT, USA 13 2Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, CT, USA 14 3Section of Plastic and Reconstructive Surgery, Department of Surgery, Yale University School of Medicine, New Haven, CT, USA 15 4Department of Biostatistics, Yale University School of Medicine, New Haven, CT, USA 16 5Yale Center for Genome Analysis, New Haven, CT, USA 17 6Craniosynostosis and Positional Plagiocephaly Support, New York, NY, USA 18 7Department of Neurosurgery, Yale University School of Medicine, New Haven, CT, USA 19 8Child Study Center, Yale University School of Medicine, New Haven, CT, USA 20 9The Rockefeller University, New York, NY, USA 21 22 ABSTRACT 23 Premature fusion of the cranial sutures (craniosynostosis), affecting 1 in 2,000 24 newborns, is treated surgically in infancy to prevent adverse neurologic outcomes. To 25 identify mutations contributing to common non-syndromic midline (sagittal and metopic) 26 craniosynostosis, we performed exome sequencing of 132 parent-offspring trios and 59 27 additional probands. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Bppart and Bpmax: RNA-RNA Interaction Partition Function and Structure Prediction for the Base Pair Counting Model
BPPart and BPMax: RNA-RNA Interaction Partition Function and Structure Prediction for the Base Pair Counting Model Ali Ebrahimpour-Boroojeny, Sanjay Rajopadhye, and Hamidreza Chitsaz ∗ Department of Computer Science, Colorado State University Abstract A few elite classes of RNA-RNA interaction (RRI), with complex roles in cellular functions such as miRNA-target and lncRNAs in human health, have already been studied. Accordingly, RRI bioinfor- matics tools tailored for those elite classes have been proposed in the last decade. Interestingly, there are somewhat unnoticed mRNA-mRNA interactions in the literature with potentially drastic biological roles. Hence, there is a need for high-throughput generic RRI bioinformatics tools. We revisit our RRI partition function algorithm, piRNA, which happens to be the most comprehensive and computationally-intensive thermodynamic model for RRI. We propose simpler models that are shown to retain the vast majority of the thermodynamic information that piRNA captures. We simplify the energy model and instead consider only weighted base pair counting to obtain BPPart for Base-pair Partition function and BPMax for Base-pair Maximization which are 225 and 1350 faster ◦ × × than piRNA, with a correlation of 0.855 and 0.836 with piRNA at 37 C on 50,500 experimentally charac- terized RRIs. This correlation increases to 0.920 and 0.904, respectively, at 180◦C. − Finally, we apply our algorithm BPPart to discover two disease-related RNAs, SNORD3D and TRAF3, and hypothesize their potential roles in Parkinson's disease and Cerebral Autosomal Dominant Arteri- opathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL). 1 Introduction Since mid 1990s with the advent of RNA interference discovery, RNA-RNA interaction (RRI) has moved to the spotlight in modern, post-genome biology. -

Datasheet Blank Template
SAN TA C RUZ BI OTEC HNOL OG Y, INC . TRIM (M-187): sc-366296 BACKGROUND APPLICATIONS TRIM (T-cell receptor interacting molecule) is a novel transmembrane adap tor TRIM (M-187) is recommended for detection of TRIM of mouse, rat and, to protein which associates and comodulates with the TCR-CD3 ζ complex in a lesser extent, human origin by Western Blotting (starting dilution 1:200, human T lymphocytes and T cell lines. TRIM is a type III transmembrane dilution range 1:100-1:1000), immunoprecipitation [1-2 µg per 100-500 µg pro tein that contains an 8-amino acid extracellular domain and an intracellu - of total protein (1 ml of cell lysate)], immunofluorescence (starting dilution lar domain that contains 4 potential phosphorylation sites and 8 tyrosine 1:50, dilution range 1:50-1:500) and solid phase ELISA (starting dilution 1:30, residues, at least 3 of which may be involved in SH2-mediated interactions dilution range 1:30-1:3000). with other signaling proteins. The human TRIM gene maps to chromosome Suitable for use as control antibody for TRIM siRNA (h): sc-106637, TRIM 3q13.13, which is a susceptibility locus for rheumatoid arthritis and is in siRNA (m): sc-154641, TRIM shRNA Plasmid (h): sc-106637-SH, TRIM shRNA proximity to the CD28, CD86, and CD80 genes, all of which encode T-cell Plasmid (m): sc-154641-SH, TRIM shRNA (h) Lentiviral Particles: sc-106637-V costimulatory molecules. TRIM is expressed in T-cells and natural killer cells, and TRIM shRNA (m) Lentiviral Particles: sc-154641-V. -

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

Producing T Cells
Lnk/Sh2b3 Controls the Production and Function of Dendritic Cells and Regulates the Induction of IFN- −γ Producing T Cells This information is current as Taizo Mori, Yukiko Iwasaki, Yoichi Seki, Masanori Iseki, of September 28, 2021. Hiroko Katayama, Kazuhiko Yamamoto, Kiyoshi Takatsu and Satoshi Takaki J Immunol published online 14 July 2014 http://www.jimmunol.org/content/early/2014/07/13/jimmun ol.1303243 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2014/07/14/jimmunol.130324 Material 3.DCSupplemental http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 28, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2014 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published July 14, 2014, doi:10.4049/jimmunol.1303243 The Journal of Immunology Lnk/Sh2b3 Controls the Production and Function of Dendritic Cells and Regulates the Induction of IFN-g–Producing T Cells Taizo Mori,*,1 Yukiko Iwasaki,*,†,1 Yoichi Seki,* Masanori Iseki,* Hiroko Katayama,* Kazuhiko Yamamoto,† Kiyoshi Takatsu,‡,x and Satoshi Takaki* Dendritic cells (DCs) are proficient APCs that play crucial roles in the immune responses to various Ags and pathogens and polarize Th cell immune responses. -

Building the Vertebrate Codex Using the Gene Breaking Protein Trap Library
Genetics, Development and Cell Biology Publications Genetics, Development and Cell Biology 2020 Building the vertebrate codex using the gene breaking protein trap library Noriko Ichino Mayo Clinic Gaurav K. Varshney National Institutes of Health Ying Wang Iowa State University Hsin-kai Liao Iowa State University Maura McGrail Iowa State University, [email protected] See next page for additional authors Follow this and additional works at: https://lib.dr.iastate.edu/gdcb_las_pubs Part of the Molecular Genetics Commons, and the Research Methods in Life Sciences Commons The complete bibliographic information for this item can be found at https://lib.dr.iastate.edu/ gdcb_las_pubs/261. For information on how to cite this item, please visit http://lib.dr.iastate.edu/howtocite.html. This Article is brought to you for free and open access by the Genetics, Development and Cell Biology at Iowa State University Digital Repository. It has been accepted for inclusion in Genetics, Development and Cell Biology Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Building the vertebrate codex using the gene breaking protein trap library Abstract One key bottleneck in understanding the human genome is the relative under-characterization of 90% of protein coding regions. We report a collection of 1200 transgenic zebrafish strains made with the gene- break transposon (GBT) protein trap to simultaneously report and reversibly knockdown the tagged genes. Protein trap-associated mRFP expression shows previously undocumented expression of 35% and 90% of cloned genes at 2 and 4 days post-fertilization, respectively. Further, investigated alleles regularly show 99% gene-specific mRNA knockdown. -

Comprehensive Array CGH of Normal Karyotype Myelodysplastic
Leukemia (2011) 25, 387–399 & 2011 Macmillan Publishers Limited All rights reserved 0887-6924/11 www.nature.com/leu LEADING ARTICLE Comprehensive array CGH of normal karyotype myelodysplastic syndromes reveals hidden recurrent and individual genomic copy number alterations with prognostic relevance A Thiel1, M Beier1, D Ingenhag1, K Servan1, M Hein1, V Moeller1, B Betz1, B Hildebrandt1, C Evers1,3, U Germing2 and B Royer-Pokora1 1Institute of Human Genetics and Anthropology, Medical Faculty, Heinrich Heine University, Duesseldorf, Germany and 2Department of Hematology, Oncology and Clinical Immunology, Heinrich Heine University, Duesseldorf, Germany About 40% of patients with myelodysplastic syndromes (MDSs) 40–50% of MDS cases have a normal karyotype. MDS patients present with a normal karyotype, and they are facing different with a normal karyotype and low-risk clinical parameters are courses of disease. To advance the biological understanding often assigned into the IPSS low and intermediate-1 risk groups. and to find molecular prognostic markers, we performed a high- resolution oligonucleotide array study of 107 MDS patients In the absence of genetic or biological markers, prognostic (French American British) with a normal karyotype and clinical stratification of these patients is difficult. To better prognosticate follow-up through the Duesseldorf MDS registry. Recurrent these patients, new parameters to identify patients at higher risk hidden deletions overlapping with known cytogenetic aberra- are urgently needed. With the more recently introduced modern tions or sites of known tumor-associated genes were identi- technologies of whole-genome-wide surveys of genetic aberra- fied in 4q24 (TET2, 2x), 5q31.2 (2x), 7q22.1 (3x) and 21q22.12 tions, it is hoped that more insights into the biology of disease (RUNX1, 2x).