Approach to Bleeding Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Rare Coagulation Disorders
Treatment OF HEMOPHILIA April 2006 · No. 39 THE RARE COAGULATION DISORDERS Paula HB Bolton-Maggs Department of Haematology Manchester Royal Infirmary Manchester, United Kingdom Published by the World Federation of Hemophilia (WFH) © World Federation of Hemophilia, 2006 The WFH encourages redistribution of its publications for educational purposes by not-for-profit hemophilia organizations. In order to obtain permission to reprint, redistribute, or translate this publication, please contact the Communications Department at the address below. This publication is accessible from the World Federation of Hemophilia’s web site at www.wfh.org. Additional copies are also available from the WFH at: World Federation of Hemophilia 1425 René Lévesque Boulevard West, Suite 1010 Montréal, Québec H3G 1T7 CANADA Tel. : (514) 875-7944 Fax : (514) 875-8916 E-mail: [email protected] Internet: www.wfh.org The Treatment of Hemophilia series is intended to provide general information on the treatment and management of hemophilia. The World Federation of Hemophilia does not engage in the practice of medicine and under no circumstances recommends particular treatment for specific individuals. Dose schedules and other treatment regimes are continually revised and new side effects recognized. WFH makes no representation, express or implied, that drug doses or other treatment recommendations in this publication are correct. For these reasons it is strongly recommended that individuals seek the advice of a medical adviser and/or to consult printed instructions provided by the pharmaceutical company before administering any of the drugs referred to in this monograph. Statements and opinions expressed here do not necessarily represent the opinions, policies, or recommendations of the World Federation of Hemophilia, its Executive Committee, or its staff. -

Section 8: Hematology CHAPTER 47: ANEMIA
Section 8: Hematology CHAPTER 47: ANEMIA Q.1. A 56-year-old man presents with symptoms of severe dyspnea on exertion and fatigue. His laboratory values are as follows: Hemoglobin 6.0 g/dL (normal: 12–15 g/dL) Hematocrit 18% (normal: 36%–46%) RBC count 2 million/L (normal: 4–5.2 million/L) Reticulocyte count 3% (normal: 0.5%–1.5%) Which of the following caused this man’s anemia? A. Decreased red cell production B. Increased red cell destruction C. Acute blood loss (hemorrhage) D. There is insufficient information to make a determination Answer: A. This man presents with anemia and an elevated reticulocyte count which seems to suggest a hemolytic process. His reticulocyte count, however, has not been corrected for the degree of anemia he displays. This can be done by calculating his corrected reticulocyte count ([3% × (18%/45%)] = 1.2%), which is less than 2 and thus suggestive of a hypoproliferative process (decreased red cell production). Q.2. A 25-year-old man with pancytopenia undergoes bone marrow aspiration and biopsy, which reveals profound hypocellularity and virtual absence of hematopoietic cells. Cytogenetic analysis of the bone marrow does not reveal any abnormalities. Despite red blood cell and platelet transfusions, his pancytopenia worsens. Histocompatibility testing of his only sister fails to reveal a match. What would be the most appropriate course of therapy? A. Antithymocyte globulin, cyclosporine, and prednisone B. Prednisone alone C. Supportive therapy with chronic blood and platelet transfusions only D. Methotrexate and prednisone E. Bone marrow transplant Answer: A. Although supportive care with transfusions is necessary for treating this patient with aplastic anemia, most cases are not self-limited. -
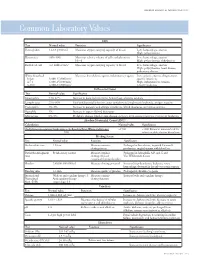
Common Laboratory Values
AmericAn AcAdemy of PediAtric dentistry Reference Manual 2006-2007 Resource Section 251 Common LaboratoryCommon Values Laboratory Values CBC Test Normal value Function Significance Hemoglobin 12-18 g/100 mL Measures oxygen carrying capacity of blood Low: hemorrhage, anemia High: polycythemia Hematocrit 35%-50% Measures relative volume of cells and plasma in Low: hemorrhage, anemia blood High: polycythemia, dehydration Red blood cell 4-6 million/mm3 Measures oxygen-carrying capacity of blood Low: hemorrhage, anemia High: polycythemia, heart disease, pulmonary disease White blood cell Measures host defense against inflammatory agents Low: aplastic anemia, drug toxicity, Infant 8,000-15,000/mm3 specific infections 4-7 y 6,000-15,000/mm3 High: inflammation, trauma, 8-18 y 4,500-13,500/mm3 toxicity, leukemia Differential Count Test Normal value Significance Neutrophils 54%-62% Increase in bacterial infections, hemorrhage, diabetic acidosis Lymphocytes 25%-30% Viral and bacterial infection, acute and chronic lymphocytic leukemia, antigen reaction Eosinophils 1%-3% Increase in parasitic and allergic conditions, blood dyscrasias, pernicious anemia Basophils 1% Increase in types of blood dyscrasias Monocytes 0%-9% Hodgkin’s disease, lipid storage disease, recovery from severe infections, monocytic leukemia Absolute Neutrophil Count (ANC) Calculation Normal value Significance (% Polymorphonuclear Leukocytes + % Bands)×Total White Cell Count >1500 <1000 Patient at increased risk for 100 infection; defer elective dental care Bleeding Screen Test -

Prolonged Partial Thromboplastin Time Without Bleeding History; Fletcher Factor Deficiency
Prolonged Partial Thromboplastin Time Without Bleeding History; Fletcher Factor Deficiency Celalettin ÜSTÜN*, Anand JILLELLA*, Linda HENDRIKS*, Mary JONAH**, Ferdane KUTLAR*, Russell BURGESS*, Abdullah KUTLAR* * Section of Hematology Oncology, Department of Medicine, Medical College of Georgia, ** Department of Pathology, Medical College of Georgia, Augusta, USA ABSTRACT A 67-year-old patient was admitted to the hospital to perform an esophagogastrectomy because a lesion at the lower esophagus was strongly suspicious for cancer. Her medical history and her family his- tory were negative for bleeding tendency or thrombosis. Her activated partial thromboplastin time (aPTT) was prolonged (44 s) whereas her prothrombin time (PT) was normal (11 s) presurgery. Mixing of her plasma with normal plasma corrected her prolonged aPTT (27.9 s). Prolonged incubation shortened the patient’s aPTT (36.3 s). Fletcher factor activity was found to be 50%. The patient underwent an esopha- gogastrectomy without bleeding complications under spinal anesthesia. Fletcher factor deficiency, a ra- re disorder, should be considered in patients who have no history of bleeding tendency with a prolonged aPTT. Surgical interventions are safe in these patients. Key Words: aPTT, Surgery, Fletcher factor, Prekallikrein. Turk J Haematol 2002;19(3): 417-419 Received: 20.06.2001 Accepted: 28.07.2001 INTRODUCTION unt[2]. Hematologists are frequently consulted for preoperative coagulation abnormalities. Preoperative evaluation of hemostasis is cru- cial to assess the risk of per-and peri-operative Prolonged aPTT is not an uncommon abnor- bleeding. The most effective screening method is mality encountered during preoperative evaluati- to obtain a thorough history of bleeding[1]. on. It may indicate the presence of antiphospholi- Preoperative screening tests mostly include acti- pid antibodies or a factor deficiency in the intrinsic vated partial thromboplastin time (aPTT), proth- and/or common pathways of blood coagulation. -

Molecular Basis of Fibrinogen Naples Associated with Defective Thrombin Binding and Thrombophilia
Molecular basis of fibrinogen Naples associated with defective thrombin binding and thrombophilia. Homozygous substitution of B beta 68 Ala----Thr. J Koopman, … , J Grimbergen, P M Mannucci J Clin Invest. 1992;90(1):238-244. https://doi.org/10.1172/JCI115841. Research Article In an abnormal fibrinogen (fibrinogen Naples) associated with congenital thrombophilia we have identified a single base substitution (G----A) in the B beta chain gene that results in an amino acid substitution of alanine by threonine at position 68 in the B beta chain of fibrinogen. The propositus and two siblings were found to be homozygous for the mutation, whereas the parents and another sibling were found to be heterozygous. Individuals homozygous for the defect had a severe history of both arterial and venous thrombosis; heterozygous individuals had no clinical symptoms. The three homozygotes had a prolonged thrombin clotting time in plasma, whereas the heterozygotes had a normal thrombin clotting time. Fibrinopeptide A and B (FpA and FpB) release from purified fibrinogen by human alpha-thrombin was delayed in both the homozygous propositus and a heterozygous family member. Release of FpA from the normal and abnormal amino-terminal disulfide knot (NDSK) corresponded to that found with the intact fibrinogens, indicating a decreased interaction of thrombin with the NDSK part of fibrinogen Naples. Binding studies showed that fibrin from homozygous abnormal fibrinogen bound less than 10% of active site inhibited alpha-thrombin as compared with normal fibrin, while fibrin formed from heterozygous abnormal fibrinogen bound approximately 50% of alpha-thrombin. These results suggest that the mutation of B beta Ala 68----Thr affects the binding of alpha-thrombin […] Find the latest version: https://jci.me/115841/pdf Molecular Basis of Fibrinogen Naples Associated with Defective Thrombin Binding and Thrombophilia Homozygous Substitution of BB 68 Ala -- Thr Jaap Koopman,** Frits Haverkate,t Susan T. -

MGH Clinical Laboratories
MGH Laboratory Handbook Online Lab Handbook: http://mghlabtest.partners.org Reference Intervals - MGH Clinical Laboratories MGH Department of Pathology 55 Fruit Street, GRJ 220 Boston, MA 02114-2696 Report generated: August 19, 2009 Test name Reference Interval Laboratory 1-25-OH Vitamin D Core lab (Sendouts) 1-3-Beta D glucan assay Core lab (Sendouts) 17-OH Progesterone Core lab (Sendouts) 25-OH Vitamin D Desired: > 32 ng/mL Core 3-Methylhistidine, urine Age matched reference range and interpretation Neurochemistry provided 5-Nucleotidase Core lab (Sendouts) A1AT deficiency profile Core lab (Sendouts) ABO/Rh Type Blood Transfusion Service Acetaminophen Negative Core Acetone Negative Core Acetylaspartate, urine Interpretation provided Neurochemistry ACTH 6-76 pg/ml Core Acylcarnitines (plasma) Core lab (Sendouts) ADAMTS13 activity/inhibitor Core lab (Sendouts) Adenosine deaminase (fluid--NOT CSF) Core lab (Sendouts) Adenovirus antibody Reported with results Microbiology Adenovirus antigen Negative Microbiology Adenylosuccinase deficiency, screen Interpretation provided Neurochemistry AFP (non-maternal specimens) Core Alanine, CSF Interpretation and age-matched reference ranges Neurochemistry provided. Wednesday, August 19, 2009 Page 1 of 26 Test name Reference Interval Laboratory Alanine, plasma Interpretation and age-matched reference ranges Neurochemistry provided. Albumin 3.1-4.3 g/dl Chelsea Healthcenter Albumin 3.3-5.0 g/dl Core Albumin (fluid--NOT CSF) Core Alcohols (ethanol, MeOH, isoprop) Negative Core Aldolase Core lab (Sendouts) -

The Role of the Laboratory in Treatment with New Oral Anticoagulants
Journal of Thrombosis and Haemostasis, 11 (Suppl. 1): 122–128 DOI: 10.1111/jth.12227 INVITED REVIEW The role of the laboratory in treatment with new oral anticoagulants T. BAGLIN Department of Haematology, Addenbrooke’s Hospital, Cambridge University Hospitals NHS Trust, Cambridge, UK To cite this article: Baglin T. The role of the laboratory in treatment with new oral anticoagulants. J Thromb Haemost 2013; 11 (Suppl. 1): 122–8. tion of thromboembolism in patients with atrial fibrilla- Summary. Orally active small molecules that selectively tion. For some patients, these drugs offer substantial and specifically inhibit coagulation serine proteases have benefits over oral vitamin K antagonists (VKAs). For the been developed for clinical use. Dabigatran etexilate, majority of patients, these drugs are prescribed at fixed rivaroxaban and apixaban are given at fixed doses and doses without the need for monitoring or dose adjustment. do not require monitoring. In most circumstances, these There are no food interactions and very limited drug inter- drugs have predictable bioavailability, pharmacokinetic actions. The rapid onset of anticoagulation and short half- effects, and pharmacodynamic effects. However, there life make the initiation and interruption of anticoagulant will be clinical circumstances when assessment of the therapy considerably easier than with VKAs. As with all anticoagulant effect of these drugs will be required. The anticoagulants produced so far, there is a correlation effect of these drugs on laboratory tests has been deter- between intensity of anticoagulation and bleeding. Conse- mined in vitro by spiking normal samples with a known quently, the need to consider the balance of benefit and risk concentration of active compound, or ex vivo by using in each individual patient is no less important than with plasma samples from volunteers and patients. -

ISTH Couverture 6.6.2012 10:21 Page 1 ISTH Couverture 6.6.2012 10:21 Page 2 ISTH Couverture 6.6.2012 10:21 Page 3 ISTH Couverture 6.6.2012 10:21 Page 4
ISTH Couverture 6.6.2012 10:21 Page 1 ISTH Couverture 6.6.2012 10:21 Page 2 ISTH Couverture 6.6.2012 10:21 Page 3 ISTH Couverture 6.6.2012 10:21 Page 4 ISTH 2012 11.6.2012 14:46 Page 1 Table of Contents 3 Welcome Message from the Meeting President 3 Welcome Message from ISTH Council Chairman 4 Welcome Message from SSC Chairman 5 Committees 7 ISTH Future Meetings Calendar 8 Meeting Sponsors 9 Awards and Grants 2012 12 General Information 20 Programme at a Glance 21 Day by Day Scientific Schedule & Programme 22 Detailed Programme Tuesday, 26 June 2012 25 Detailed Programme Wednesday, 27 June 2012 33 Detailed Programme Thursday, 28 June 2012 44 Detailed Programme Friday, 29 June 2012 56 Detailed Programme Saturday, 30 June 2012 68 Hot Topics Schedule 71 ePoster Sessions 97 Sponsor & Exhibitor Profiles 110 Exhibition Floor Plan 111 Congress Centre Floor Plan www.isth.org ISTH 2012 11.6.2012 14:46 Page 2 ISTH 2012 11.6.2012 14:46 Page 3 WelcomeCommittees Messages Message from the ISTH SSC 2012 Message from the ISTH Meeting President Chairman of Council Messages Dear Colleagues and Friends, Dear Colleagues and Friends, We warmly welcome you to the elcome It is my distinct privilege to welcome W Scientific and Standardization Com- you to Liverpool for our 2012 SSC mittee (SSC) meeting of the Inter- meeting. national Society on Thrombosis and Dr. Cheng-Hock Toh and his col- Haemostasis (ISTH) at Liverpool’s leagues have set up a great Pro- UNESCO World Heritage Centre waterfront! gramme aiming at making our off-congress year As setting standards is fundamental to all quality meeting especially attractive for our participants. -

Whole-Exome Sequencing of a Patient with Severe and Complex Hemostatic Abnormalities Reveals a Possible Contributing Frameshift Mutation in C3AR1
Downloaded from molecularcasestudies.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Whole-exome sequencing of a patient with severe and complex hemostatic abnormalities reveals a possible contributing frameshift mutation in C3AR1 Eva Leinøe1, Ove Juul Nielsen1, Lars Jønson2 and Maria Rossing2∗ Department of Hematology1 and Center for Genomic Medicine2, Rigshospitalet, University of Copenhagen, Blegdamsvej 9, DK-2100 Copenhagen, Denmark Running head: WES reveals a C3AR1 mutation in a complex hemostatic patient ∗Corresponding author: Maria Rossing Center for Genomic Medicine Rigshospitalet University of Copenhagen Blegdamsvej 9 DK-2100 Copenhagen Denmark E-mail: [email protected] Phone: +45 3545 3016 Fax: +45 3545 4435 1 Downloaded from molecularcasestudies.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract The increasing availability of genome-wide analysis has made it possible to rapidly sequence the exome of patients with undiagnosed or unresolved medical conditions. Here, we present the case of a 64-year-old male patient with schistocytes in the peripheral blood smear and a complex and life-threatening coagulation disorder causing recurrent venous thromboembolic events, severe thrombocytopenia, and subdural hematomas. Whole-exome sequencing revealed a frameshift mutation (C3AR1 c.355-356dup, p.Asp119Alafs*19) resulting in a premature stop in C3AR1 (Complement Component 3a Receptor 1). Based on this finding, atypical hemolytic uremic syndrome was suspected due to a genetic predisposition, and a targeted treatment regime with Eculizumab was initiated. Life-threatening hemostatic abnormalities would most likely have persisted had it not been for the implementation of whole-exome sequencing in this particular clinical setting. -

Approach to Coagulopathy in the Icu Dic and Thrombotic Emergencies
APPROACH TO COAGULOPATHY IN THE ICU DIC AND THROMBOTIC EMERGENCIES NEIL KUMAR, MD UNIVERSITY OF ROCHESTER MEDICAL CENTER Disclosures u I have no financial disclosures u I am NOT A HEMATOLOGIST Outline u Review of hemostasis and coagulopathy u Discuss laboratory markers for coagulopathy u Discuss an approach to a few specific coagulopathies and thrombotic emergencies Outline u Review of hemostasis and coagulopathy u Discuss laboratory markers for coagulopathy u Discuss an approach to a few specific coagulopathies and thrombotic emergencies Coagulation u Coagulation is the process in which blood clots u Fibrinolysis is the process in which clot dissolves u Hemostasis is the stopping of bleeding or hemorrhage. u Ideally, hemostasis is a balance between coagulation and fibrinolysis Coagulation (classic pathways) Michael G. Crooks Simon P. Hart Eur Respir Rev 2015;24:392-399 Coagulation (another view) Gando, S. et al. (2016) Disseminated intravascular coagulation Nat. Rev. Dis. Primers doi:10.1038/nrdp.2016.37 Coagulation (yet another view) u Inflammation and coagulation intersect with platelets in the middle u An example of this is Disseminated Intravascular Coagulation. Gando, S. et al. (2016) Disseminated intravascular coagulation Nat. Rev. Dis. Primers doi:10.1038/nrdp.2016.37 Outline u Review of hemostasis and coagulopathy u Discuss laboratory markers for coagulopathy u Discuss an approach to a few specific coagulopathies and thrombotic emergencies PT / INR u Prothrombin Time u Test of Extrinsic Pathway u Take plasma (blood without cells) and re-add calcium u Calcium was removed with citrate in tube u Add tissue factor u See how long it takes to clot and normalize PT to get INR Coagulation (classic pathways) Michael G. -

Diagnosis of Hemophilia and Other Bleeding Disorders
Diagnosis of Hemophilia and Other Bleeding Disorders A LABORATORY MANUAL Second Edition Steve Kitchen Angus McCraw Marión Echenagucia Published by the World Federation of Hemophilia (WFH) © World Federation of Hemophilia, 2010 The WFH encourages redistribution of its publications for educational purposes by not-for-profit hemophilia organizations. For permission to reproduce or translate this document, please contact the Communications Department at the address below. This publication is accessible from the World Federation of Hemophilia’s website at www.wfh.org. Additional copies are also available from the WFH at: World Federation of Hemophilia 1425 René Lévesque Boulevard West, Suite 1010 Montréal, Québec H3G 1T7 CANADA Tel.: (514) 875-7944 Fax: (514) 875-8916 E-mail: [email protected] Internet: www.wfh.org Diagnosis of Hemophilia and Other Bleeding Disorders A LABORATORY MANUAL Second Edition (2010) Steve Kitchen Angus McCraw Marión Echenagucia WFH Laboratory WFH Laboratory (co-author, Automation) Training Specialist Training Specialist Banco Municipal Sheffield Haemophilia Katharine Dormandy de Sangre del D.C. and Thrombosis Centre Haemophilia Centre Universidad Central Royal Hallamshire and Thrombosis Unit de Venezuela Hospital The Royal Free Hospital Caracas, Venezuela Sheffield, U.K. London, U.K. on behalf of The WFH Laboratory Sciences Committee Chair (2010): Steve Kitchen, Sheffield, U.K. Deputy Chair: Sukesh Nair, Vellore, India This edition was reviewed by the following, who at the time of writing were members of the World Federation of Hemophilia Laboratory Sciences Committee: Mansoor Ahmed Clarence Lam Norma de Bosch Sukesh Nair Ampaiwan Chuansumrit Alison Street Marión Echenagucia Alok Srivastava Andreas Hillarp Some sections were also reviewed by members of the World Federation of Hemophilia von Willebrand Disease and Rare Bleeding Disorders Committee. -

Approach to Bleeding Diathesi
Approach to Bleeding Diathesis Dr.Nalini K Pati MD, DNB, DCH (Syd), FRCPA Paediatric Haematologist Royal Children’s Hospital Melbourne Australia Objectives Objectives - I I. Clinical aspects of bleeding Clinical aspects of bleeding II. Hematologic disorders causing bleeding • Coagulation factor disorders • Platelet disorders III. Approach to acquired bleeding disorders • Hemostasis in liver disease • Surgical patients • Warfarin toxicity IV. Approach to laboratory abnormalities • Diagnosis and management of thrombocytopenia V. Drugs and blood products used for bleeding Clinical Features of Bleeding Disorders Petechiae Platelet Coagulation (typical of platelet disorders) disorders factor disorders Site of bleeding Skin Deep in soft tissues Mucous membranes (joints, muscles) (epistaxis, gum, vaginal, GI tract) Petechiae Yes No Ecchymoses (“bruises”) Small, superficial Large, deep Hemarthrosis / muscle bleeding Extremely rare Common Do not blanch with pressure Bleeding after cuts & scratches Yes No (cf. angiomas) Bleeding after surgery or trauma Immediate, Delayed (1-2 days), usually mild often severe Not palpable (cf. vasculitis) Ecchymoses (typical of coagulation factor disorders) Objectives - II Hematologic disorders causing bleeding – Coagulation factor disorders – Platelet disorders Coagulation factor disorders Hemophilia A and B Inherited bleeding Acquired bleeding Hemophilia A Hemophilia B disorders disorders Coagulation factor deficiency Factor VIII Factor IX – Hemophilia A and B – Liver disease – vonWillebrands disease – Vitamin K Inheritance X-linked X-linked recessive recessive – Other factor deficiencies deficiency/warfarin overdose Incidence 1/10,000 males 1/50,000 males –DIC Severity Related to factor level <1% - Severe - spontaneous bleeding 1-5% - Moderate - bleeding with mild injury 5-25% - Mild - bleeding with surgery or trauma Complications Soft tissue bleeding Hemarthrosis (acute) Hemophilia Clinical manifestations (hemophilia A & B are indistinguishable) Hemarthrosis (most common) Fixed joints Soft tissue hematomas (e.