Severity of Anaemia Has Corresponding Effects on Coagulation Parameters of Sickle Cell Disease Patients
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
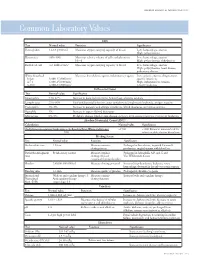
Common Laboratory Values
AmericAn AcAdemy of PediAtric dentistry Reference Manual 2006-2007 Resource Section 251 Common LaboratoryCommon Values Laboratory Values CBC Test Normal value Function Significance Hemoglobin 12-18 g/100 mL Measures oxygen carrying capacity of blood Low: hemorrhage, anemia High: polycythemia Hematocrit 35%-50% Measures relative volume of cells and plasma in Low: hemorrhage, anemia blood High: polycythemia, dehydration Red blood cell 4-6 million/mm3 Measures oxygen-carrying capacity of blood Low: hemorrhage, anemia High: polycythemia, heart disease, pulmonary disease White blood cell Measures host defense against inflammatory agents Low: aplastic anemia, drug toxicity, Infant 8,000-15,000/mm3 specific infections 4-7 y 6,000-15,000/mm3 High: inflammation, trauma, 8-18 y 4,500-13,500/mm3 toxicity, leukemia Differential Count Test Normal value Significance Neutrophils 54%-62% Increase in bacterial infections, hemorrhage, diabetic acidosis Lymphocytes 25%-30% Viral and bacterial infection, acute and chronic lymphocytic leukemia, antigen reaction Eosinophils 1%-3% Increase in parasitic and allergic conditions, blood dyscrasias, pernicious anemia Basophils 1% Increase in types of blood dyscrasias Monocytes 0%-9% Hodgkin’s disease, lipid storage disease, recovery from severe infections, monocytic leukemia Absolute Neutrophil Count (ANC) Calculation Normal value Significance (% Polymorphonuclear Leukocytes + % Bands)×Total White Cell Count >1500 <1000 Patient at increased risk for 100 infection; defer elective dental care Bleeding Screen Test -

Prothrombin Time (PT) Be Kept in Constant and Incubation Temperature at Step 3, Start Stop Watch , Mix in a Water Bath ( 37°C ) (LIQUID REAGENT) 36.5-37.5°C
components of any other kit with different lot numbers. temperature 15 minutes prior to testing. Throughout testing all test tubes, syringes and pipettes 2. Pre-warm PT reagent at 37°C for 5 minutes. should be plastic. 3. Pipette 100µl of PT reagent to each tube. Throughout testing all test tubes, incubation time should 4. Add 50 µl of sample, controls to the tubes prepared in Prothrombin Time (PT) be kept in constant and incubation temperature at step 3, start stop watch , mix in a water bath ( 37°C ) (LIQUID REAGENT) 36.5-37.5°C. for 8 seconds , then record the time required for clot For In-Vitro diagnostic use only If testing is delayed for more than 4 hours, plasma may formation . be stored at 2-8°C. B.Automated Method Store at: 2-8°C Each laboratory should establish a Quality Control To perform this test, refer to the appropriate Instrument program that includes both normal and abnormal control Operator’s Manual for detailed instructions. INTENDED USE plasmas to evaluate instrument, reagent tested daily Prothrombin Time (PT) is commonly used for screening for prior to performing tests on patient plasmas. Monthly RESULTS extrinsic factor deficiency, monitoring oral anticoagulant quality control charts are recommended to determine Clot time of the test Prothrombin Time plasma therapy and quantitative determination of the extrinsic the mean and standard deviation of each of the daily = Ratio (PTR) Clot time of the control coagulation factors. control plasma. All assays should include controls, and if plasma any of the controls are outside the established reference INR = PTRISI PRINCIPLE ranges, then the assay should be considered invalid and Example: for a PTR of 2.0 and an ISI of 1.0 Tissue thromboplastin, in the presence of calcium ions and no patient results should be reported. -

Blood and Immunity
Chapter Ten BLOOD AND IMMUNITY Chapter Contents 10 Pretest Clinical Aspects of Immunity Blood Chapter Review Immunity Case Studies Word Parts Pertaining to Blood and Immunity Crossword Puzzle Clinical Aspects of Blood Objectives After study of this chapter you should be able to: 1. Describe the composition of the blood plasma. 7. Identify and use roots pertaining to blood 2. Describe and give the functions of the three types of chemistry. blood cells. 8. List and describe the major disorders of the blood. 3. Label pictures of the blood cells. 9. List and describe the major disorders of the 4. Explain the basis of blood types. immune system. 5. Define immunity and list the possible sources of 10. Describe the major tests used to study blood. immunity. 11. Interpret abbreviations used in blood studies. 6. Identify and use roots and suffixes pertaining to the 12. Analyse several case studies involving the blood. blood and immunity. Pretest 1. The scientific name for red blood cells 5. Substances produced by immune cells that is . counteract microorganisms and other foreign 2. The scientific name for white blood cells materials are called . is . 6. A deficiency of hemoglobin results in the disorder 3. Platelets, or thrombocytes, are involved in called . 7. A neoplasm involving overgrowth of white blood 4. The white blood cells active in adaptive immunity cells is called . are the . 225 226 ♦ PART THREE / Body Systems Other 1% Proteins 8% Plasma 55% Water 91% Whole blood Leukocytes and platelets Formed 0.9% elements 45% Erythrocytes 10 99.1% Figure 10-1 Composition of whole blood. -

Research Article D-Dimer and Prothrombin Time Are the Significant Indicators of Severe COVID-19 and Poor Prognosis
Hindawi BioMed Research International Volume 2020, Article ID 6159720, 10 pages https://doi.org/10.1155/2020/6159720 Research Article D-Dimer and Prothrombin Time Are the Significant Indicators of Severe COVID-19 and Poor Prognosis Hui Long,1 Lan Nie,2 Xiaochen Xiang,2 Huan Li,1 Xiaoli Zhang,1 Xiaozhi Fu,1 Hongwei Ren,1 Wanxin Liu,2 Qiang Wang ,2 and Qingming Wu 1,2 1Internal Medicine of Tianyou Hospital, Wuhan University of Science and Technology, Wuhan 430064, China 2Institute of Infection, Immunology and Tumor Microenvironment, Hubei Province Key Laboratory of Occupational Hazard Identification and Control, Medical College, Wuhan University of Science and Technology, Wuhan 430065, China Correspondence should be addressed to Qiang Wang; [email protected] and Qingming Wu; [email protected] Received 22 April 2020; Accepted 19 May 2020; Published 17 June 2020 Academic Editor: Frederick D. Quinn Copyright © 2020 Hui Long et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Objective. To investigate the value of coagulation indicators D-dimer (DD), prothrombin time (PT), activated partial thromboplastin time (APTT), thrombin time (TT), and fibrinogen (Fg) in predicting the severity and prognosis of COVID-19. Methods. A total of 115 patients with confirmed COVID-19, who were admitted to Tianyou Hospital of Wuhan University of Science and Technology between January 18, 2020, and March 5, 2020, were included. The dynamic changes of DD, PT, APTT, and Fg were tested, and the correlation with CT imaging, clinical classifications, and prognosis was studied. -

Hemostasis and Thrombosis
PROCEDURES FOR HEMOSTASIS AND THROMBOSIS A Clinical Test Compendium PROCEDURES FOR HEMOSTASIS AND THROMBOSIS: A CLINICAL TEST COMPENDIUM Test No. Test Name Profile Includes Specimen Requirements Bleeding Profiles and Screening Tests 117199 aPTT Mixing Studies aPTT; aPTT 1:1 mix normal plasma (NP); aPTT 1:1 mix saline; aPTT 2 mL citrated plasma, frozen 1:1 mix, incubated; aPTT 1:1 mix NP, incubated control 116004 Abnormal Bleeding Profile PT; aPTT; thrombin time; platelet count 5 mL EDTA whole blood, one tube citrated whole blood (unopened), and 2 mL citrated plasma, frozen Minimum: 5 mL EDTA whole blood, one tube citrated whole blood (unopened), and 1 mL citrated plasma, frozen 503541 Bleeding Diathesis With Normal α2-Antiplasmin assay; euglobulin lysis time; factor VIII activity; 7 mL (1mL in each of 7 tubes) platelet-poor aPTT/PT Profile (Esoterix) factor VIII chromogenic; factor IX activity; factor XI activity; factor citrated plasma, frozen XIII activity; fibrinogen activity; PAI-1 activity with reflex to PAI-1 antigen and tPA; von Willebrand factor activity; von Willebrand factor antigen 336572 Menorrhagia Profile PT; aPTT; factor IX activity; factor VIII activity; factor XI activity; 3 mL citrated plasma, frozen von Willebrand factor activity; von Willebrand factor antigen Minimum: 2 mL citrated plasma, frozen 117866 Prolonged Protime Profile Factor II activity; factor V activity; factor VII activity; factor 3 mL citrated plasma, frozen X activity; fibrinogen activity; dilute prothrombin time Minimum: 2 mL citrated plasma, frozen -

Thrombophilia Testing Panel Now Available
Thrombophilia Testing Panel Now Available Bronson Laboratory offers a any impact on the patient’s therapy. Importantly, most people with an thrombophilia testing panel consisting For instance, the vast majority of older inheritable thrombophilic mutation will of the following: factor VIII activity, anti- patients with venous thromboses in the never experience a thrombotic event, thrombin III activity, protein setting of a recognized inciting event and most of those who do will never C activity, protein S antigen, factor V Leiden will not have a preexisting thrombophilic do so again. Because of this, screening mutation assay, prothrombin 90210A condition, and accordingly testing is the general population or even asympto- mutation assay, and a lupus anticoagulant generally not recommended in this pop- matic relatives of individuals with thrombo- assay. Abnormal values for protein C, ulation. In regards to arterial thromboses, philic mutations is discouraged because protein S, and antithrombin III will be the overwhelming majority are a direct of the potential for harmful overtreatment. automatically reflexed to confirmatory consequence of atherosclerosis, and the By similar reasoning, some hematologists testing at the Mayo Clinic laboratories. added presence of a thrombophilia in would recommend the same period of Although these tests are orderable indi- this setting will not affect patient anticoagulation, often 3-6 months after vidually, there is little rationale to do so management. (continued on page 4) except as noted below. Clinical circumstances that suggest The activity assays are clotting tests a laboratory evaluation for thrombophilia that require adequate amounts of func- tional protein to achieve a normal result. For reasons having to do with greater achievable precision, the free protein S Patient with a Asymptomatic patients Other clinical situations with a family history of VTE antigen test is used in lieu of an activity confirmed acute VTE assay, but it too depends on adequate amounts of circulating protein for a normal finding. -

HEMOCLOT™ Thrombin Time
HEMOCLOT™ Thrombin Time (T.T.) REF CK011K R 6 x 2 mL Sales and Support: CoaChrom Diagnostica GmbH REF CK011L R 6 x 8 mL www.coachrom.com | [email protected] Tel: +43-1-236 222 1 | Fax: +43-1-236 222 111 Toll-free contact for Germany: Tel: 0800-24 66 33-0 | Fax: 0800-24 66 33-3 Clotting method for the determination of Thrombin Time. English, last revision: 08-2019 INTENDED USE: R Reconstitute the contents of each vial with exactly: HEMOCLOT™ Thrombin Time (T.T.) kit is a clotting method for the in REF CK011K ➔ 2 mL of distilled water. vitro qualitative determination of Thrombin Time on human citrated REF CK011L ➔ 8 mL of distilled water. plasma, using manual or automated method. Shake vigorously until complete dissolution while avoiding formation of SUMMARY AND EXPLANATION: foam and load it directly on the analyzer following application guide Technical: instruction. The Thrombin Time is a coagulation assay measuring the time to For manual method, allow to stabilize for 15 minutes at room convert fibrinogen to fibrin. temperature (18-25°C), homogenize before use. Clinical:1 The Thrombin Time is a screening test to assess abnormalities of STORAGE AND STABILITY: fibrinogen and to detect inhibitors against thrombin or fibrin. It can be Unopened reagents should be stored at 2-8°C in their original useful for the evaluation of Disseminated Intravascular Coagulation packaging. Under these conditions, they can be used until the expiry (DIC) and liver disease. date printed on the kit. A prolonged Thrombin Time can result from: • Presence of antithrombin activity induced by therapy (eg Heparin, R Reagent stability after reconstitution, free from any contamination hirudin, argatroban, dabigatran). -

Association Between Coagulation Function and Patients with Primary Angle Closure Glaucoma: a 5-Year Retrospective Case–Control Study
Open Access Research BMJ Open: first published as 10.1136/bmjopen-2017-016719 on 4 November 2017. Downloaded from Association between coagulation function and patients with primary angle closure glaucoma: a 5-year retrospective case–control study Shengjie Li,1 Yanting Gao,1 Mingxi Shao,1 Binghua Tang,1 Wenjun Cao,1,2 Xinghuai Sun2,3,4,5 To cite: Li S, Gao Y, Shao M, ABSTRACT Strengths and limitations of this study et al. Association between Objective To evaluate the association between coagulation function and coagulation function and patients with primary angle patients with primary ► This is the first study focusing on the evaluation of closure glaucoma (PACG). angle closure glaucoma: a the coagulation function in primary angle closure 5-year retrospective case– Design A retrospective, hospital-based, case–control glaucoma (PACG) and its relationship with disease control study. BMJ Open study. severity. 2017;7:e016719. doi:10.1136/ Setting Shanghai, China. ► The large number of studied subjects. bmjopen-2017-016719 Participants A total of 1778 subjects were recruited from ► The detailed demographic and clinical data of the Eye & ENT Hospital of Fudan University from January ► Prepublication history and studied subjects. additional material for this paper 2010 to December 2015, including patients with PACG ► The nature of this study was a single eye centre, are available online. To view (male=296; female=569) and control subjects (male=290; retrospective case–control study. please visit the journal (http:// female=623). ► Statistical power analysis for specificity ability was dx. doi. org/ 10. 1136/ bmjopen- Outcome measures Sociodemographic data and limited. 2017- 016719). -

Reptilase@'-R-A New Reagent in Blmd Coagulation
British Journal of Haematology, 1971, 21, 43. Reptilase@'-R-A New Reagent in Blmd Coagulation C. FUNK,J. GMUR,R. HEROLDAND P. W. STRAUB Department of Medicine, Kantonsspital, University of Zurich, Switzerland (Received 8 September 1970; accepted for piiblication 29 September 1970) SUMMARY.Reptilase@-R, a purified thrombin-like snake-venom enzyme from Bothrops atrox, has been tested for its usefulness as a substitute for thrombin in two coagulation assay systems. Reptilase was found to be more stable than thrombin and not to be inhibited by heparin and hirudin. Performance of the Reptilase time allows the rapid exclusion of heparin con- tamination in plasma samples with prolonged thrombin times. In the presence of fibrinogenlfibrin split products, the Reptilase time is less prolonged than the thrombin time, whereas in patients with prolonged thrombin times because of congenital dysfibrinogenaemia the inverse is found. Thus, performance of both Reptilase time and thrombin time allows the rapid identification of three major causes for a prolongation of the thrombin time. In patients on heparin therapy because of a defibrination syndrome, the Reptilase time may replace the thrombin time as a guide for the degree of coagulation inhibition by FDP, although our re- sults show that it is somewhat less sensitive. Reptilase can also be substituted for thrombin in the rapid chronometric fibrino- gen assay. Unfortunately the advantages of reagent stability and insensitivity to heparin are balanced by a lesser reproducibility, especially in the presence of FDP. While thrombin liberates the fibrinopeptides of both the a- and p-chains of fibrinogen, the thrombin-like snake-venom enzyme Reptilase exclusively acts on the a-chain, thus leading to coagulation by liberating only fibrinopeptide A (Blombick et al, 1957; Stocker & Straub, 1970). -

EDUCATION Four Compartments
J R Coll Physicians Edinb 2014; 44:42–5 CME http://dx.doi.org/10.4997/JRCPE.2014.110 © 2014 Royal College of Physicians of Edinburgh Coagulation YL Chee Consultant Haematologist, Department of Haematology-Oncology, National University Hospital, Singapore ABSTRACT The haemostatic system comprises four compartments: the vasculature, Correspondence to YL Chee platelets, coagulation factors, and the fibrinolytic system. There is presently no Consultant Haematologist laboratory or near-patient test capable of reproducing the complex regulated Department of Haematology- Oncology interaction between these four compartments. The prothrombin time (PT) and NUHS Tower Block Level 7 activated partial thromboplastin time (APTT) only test the coagulation protein 1E Kent Ridge Road compartment of the system and results have to be carefully interpreted in the Singapore 119228 context of the clinical presentation and assay limitations. This article will give a e-mail general overview of the limitations of PT and APTT and discuss specific issues that [email protected] need to be considered when the tests are requested, in the context of anticoagulant monitoring, bleeding symptoms, and routine preoperative screening. Of these indications, routine preoperative screening is the most controversial and is generally not warranted in the absence of an abnormal bleeding history. KEYWORDS Prothrombin time, activated partial thromboplastin time, thrombin clotting time, preoperative screening, bleeding history, coagulation screen DECLARATIONS OF INTERESTS No conflicts of interest declared. INTRODUCTION Intrinsic pathway Extrinsic pathway The normal haemostatic system comprises four Factor XII/HMWK*/PK** compartments, the vasculature, platelets, coagulation proteins and the fibrinolytic system. When a blood Factor XI Factor XIa vessel is injured, all four compartments interact in a coordinated manner to prevent blood loss by forming a Factor IX Factor IXa Factor VIIa clot and localising this to the area of injury. -

Thrombin Time
Lab Dept: Coagulation Test Name: THROMBIN TIME General Information Lab Order Codes: TT Synonyms: Fibrin Time; Fibrinogen screen, Thrombin Time, TT CPT Codes: 85670 – Thrombin Time, plasma Test Includes: Thrombin Time reported in seconds. Logistics Test Indications: Useful at some institutions for monitoring heparin therapy or aiding in the diagnosis of disseminated intravascular coagulation (DIC), fibrinolysis, and the presence of inhibitors. Lab Testing Sections: Coagulation Phone Numbers: MIN Lab: 612-813-6280 STP Lab: 651-220-6551 Test Availability: Daily, 24 hours Turnaround Time: 2 hours Special Instructions: ● Patient’s with hematocrit levels >55% must have a special tube made to adjust for the hematocrit; contact the laboratory for special tube. ● Depending on the patient's hematocrit, up to 3 assays can be determined using one 1.8 mL light blue top tube. Specimen Specimen Type: Whole blood Container: Light Blue top tube (Buffered Na Citrate 3.2%) Draw Volume: 1.8 mL or 2.7 mL blood Processed Volume: 0.9 mL (Minimum: 0.5 mL) plasma Collection: ● A clean venipuncture is essential, avoid foaming. ● Entire sample must be collected with single collection, pooling of sample is unacceptable. ● Capillary collection is unacceptable. ● Patient’s with a hematocrit level >55% must have a special tube made to adjust for the hematocrit; contact lab for a special tube. ● Mix thoroughly by gentle inversion. Deliver immediately to the laboratory at room temperature via courier or pneumatic tube. Off campus collections: ● Must be tested within 4 hours. ● Do not refrigerate. ● If not received in our lab within 4 hours of collection, sample must be centrifuged and *platelet-poor plasma removed from cells and transferred to an aliquot tube. -

Abnormal Hemostasis Screening Tests Leading to Diagnosis of Multiple Myeloma
Acta Haematologica Polonica 50(1) • March 2019 • 32-35 • DOI: 10.2478/ahp-2019-0006 CASE REPORT / KAZUISTYKA journal homepage: https://content.sciendo.com/ahp Abnormal hemostasis screening tests leading to diagnosis of multiple myeloma Article history: Abstract Received: 05.11.2018 Multiple myeloma (MM) is a rare malignancy, characterized by clonal proliferation of plasma cells, secreting monoclonal immunoglobu- Accepted: 04.02.2019 lin. It is usually diagnosed based on histopathologic and immunophenotypic bone marrow examination. Abnormal results of screening coagulation tests, including prothrombin time, activated partial thromboplastin time and thrombin time, are commonly encountered Teresa Iwaniec1*, in patients with plasma cell neoplasms. They do not, however, reflect bleeding tendency. We describe a 71-year-old patient who was Joanna Zdziarska2, 2 accidentally diagnosed with multiple myeloma during coagulation diagnostics. Artur Jurczyszyn © 2019 Polish Society of Hematology and Transfusion Medicine, Insitute of Hematology and Transfusion Medicine. All rights reserved. 1 Department of Medicine, Jagiellonian University Medical College, Krakow, Poland 2 Department of Haematology, Jagiellonian Keywords: University Medical College, Krakow, Poland multiple myleoma, paraprotein, hemostasis screening test, thrombin time Multiple myeloma (MM) is a rare malignancy, characterized by test results, including prothrombin time (PT), activated partial clonal proliferation of plasma cells. Monoclonal immunoglobulin thromboplastin time (APTT), and