Introductory Remarks on the Milieux of the Egg and the Early Embryo
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Rise and Fall of the Bovine Corpus Luteum
University of Nebraska Medical Center DigitalCommons@UNMC Theses & Dissertations Graduate Studies Spring 5-6-2017 The Rise and Fall of the Bovine Corpus Luteum Heather Talbott University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Biochemistry Commons, Molecular Biology Commons, and the Obstetrics and Gynecology Commons Recommended Citation Talbott, Heather, "The Rise and Fall of the Bovine Corpus Luteum" (2017). Theses & Dissertations. 207. https://digitalcommons.unmc.edu/etd/207 This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected]. THE RISE AND FALL OF THE BOVINE CORPUS LUTEUM by Heather Talbott A DISSERTATION Presented to the Faculty of the University of Nebraska Graduate College in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biochemistry and Molecular Biology Graduate Program Under the Supervision of Professor John S. Davis University of Nebraska Medical Center Omaha, Nebraska May, 2017 Supervisory Committee: Carol A. Casey, Ph.D. Andrea S. Cupp, Ph.D. Parmender P. Mehta, Ph.D. Justin L. Mott, Ph.D. i ACKNOWLEDGEMENTS This dissertation was supported by the Agriculture and Food Research Initiative from the USDA National Institute of Food and Agriculture (NIFA) Pre-doctoral award; University of Nebraska Medical Center Graduate Student Assistantship; University of Nebraska Medical Center Exceptional Incoming Graduate Student Award; the VA Nebraska-Western Iowa Health Care System Department of Veterans Affairs; and The Olson Center for Women’s Health, Department of Obstetrics and Gynecology, Nebraska Medical Center. -

Profiling of Transcripts and Proteins Modulated by the E7 Oncogene in the Lung Tissue of E7-Tg Mice by the Omics Approach
MOLECULAR MEDICINE REPORTS 2: 129-137, 2009 129 Profiling of transcripts and proteins modulated by the E7 oncogene in the lung tissue of E7-Tg mice by the omics approach EUNJIN KIM1*, JEONGWOO KANG1,3*, MINCHUL CHO1, SOJUNG LEE1, EUNHEE SEO1, HEESOOK CHOI1, YUMI KIM1, JUNGHEE KIM1, KUM YONG KANG2, KWANG PYO KIM2, JAEYONG HAN3, YHUNYHONG SHEEN4, YOUNG NA YUM5, SUE-NIE PARK5 and DO-YOUNG YOON1 Departments of 1Bioscience and Biotechnology, and 2Molecular Biotechnology, Konkuk University, Hwayang-dong 1, Gwangjin-gu, Seoul 143-701; 3Laboratory of Animal Genetic Engineering, Department of Food and Animal Biotechnology, Seoul National University, Seoul 151-742; 4School of Pharmacy, Ewha Womans University, Seoul 120-750; 5Korea Food and Drug Administration, #194 Tongil-ro, Eunpyung-gu, Seoul 122-704, Korea Received August 18, 2008; Accepted November 10, 2008 DOI: 10.3892/mmr_00000073 Abstract. The E6 and E7 oncoproteins of human papilloma suggest that the E7 oncogene modulates the expression levels virus (HPV) type 16 have been known to cooperatively induce of cell cycle-related (cyclin B1, cyclin E2) and cell adhesion- the immortalization and transformation of primary keratino- and migration-related (actinin ·1, CD166) factors, which may cytes. We established an E7 transgenic mouse model to play important roles in cellular transformation in cancer. In screen HPV-related biomakers using the omics approach. addition, the solubilization of the rigid intermediate filament The methods used to identify HPV-modulated factors were network by specific proteolysis mediated via up-regulating genomics analysis by microarray using the Affymetrix 430 gelsolin and down-regulating cofilin-1, as well as increased 2.0 array to screen E7-modulated genes, and proteomics levels of endoplasmic reticulum protein calnexin with chap- analysis using nano-LC-ESI-MS/MS to screen E7-modulated erone functions, might also be involved in E7-lung epithelial proteins with the lung tissue of E7 transgenic mice. -

Tumor Suppressive Microrna-375 Regulates Lactate Dehydrogenase B in Maxillary Sinus Squamous Cell Carcinoma
INTERNATIONAL JOURNAL OF ONCOLOGY 40: 185-193, 2012 Tumor suppressive microRNA-375 regulates lactate dehydrogenase B in maxillary sinus squamous cell carcinoma TAKASHI KINoshita1,2, NIjIRo Nohata1,2, HIRoFUMI YoSHINo3, ToYoYUKI HANAzAwA2, NAoKo KIKKAwA2, LISA FUjIMURA4, TAKeSHI CHIYoMARU3, KAzUMoRI KAwAKAMI3, HIDeKI eNoKIDA3, Masayuki NAKAGAwA3, Yoshitaka oKAMoTo2 and NAoHIKo SeKI1 Departments of 1Functional Genomics, 2otorhinolaryngology/Head and Neck Surgery, Chiba University Graduate School of Medicine, 1-8-1 Inohana Chuo-ku, Chiba 260-8670; 3Department of Urology, Graduate School of Medical and Dental Sciences, Kagoshima University, 8-35-1 Sakuragaoka, Kagoshima 890-8520; 4Biomedical Research Center, Chiba University, 1-8-1 Inohana Chuo-ku, Chiba 260-8670, japan Received july 6, 2011; Accepted August 23, 2011 DoI: 10.3892/ijo.2011.1196 Abstract. The expression of microRNA-375 (miR-375) is head and neck tumors, with an annual incidence of 0.5-1.0 per significantly reduced in cancer tissues of maxillary sinus squa- 100,000 people (1,2). Since the clinical symptoms of patients mous cell carcinoma (MSSCC). The aim of this study was to with MSSCC are very insidious, tumors are often diagnosed at investigate the functional significance ofmiR-375 and a possible advanced stages. Despite advances in multimodality therapy regulatory role in the MSSCC networks. Restoration of miR-375 including surgery, radiotherapy and chemotherapy, the 5-year significantly inhibited cancer cell proliferation and invasion survival rate for MSSCC has remained ~50%. Although regional in IMC-3 cells, suggesting that miR-375 functions as a tumor lymph node metastasis and distant metastasis are uncommon suppressor in MSSCC. Genome-wide gene expression data and (20%), the high rate of locoregional recurrence (60%) contributes luciferase reporter assays indicated that lactate dehydro genase B to poor survival (3). -

Calreticulin—Multifunctional Chaperone in Immunogenic Cell Death: Potential Significance As a Prognostic Biomarker in Ovarian
cells Review Calreticulin—Multifunctional Chaperone in Immunogenic Cell Death: Potential Significance as a Prognostic Biomarker in Ovarian Cancer Patients Michal Kielbik *, Izabela Szulc-Kielbik and Magdalena Klink Institute of Medical Biology, Polish Academy of Sciences, 106 Lodowa Str., 93-232 Lodz, Poland; [email protected] (I.S.-K.); [email protected] (M.K.) * Correspondence: [email protected]; Tel.: +48-42-27-23-636 Abstract: Immunogenic cell death (ICD) is a type of death, which has the hallmarks of necroptosis and apoptosis, and is best characterized in malignant diseases. Chemotherapeutics, radiotherapy and photodynamic therapy induce intracellular stress response pathways in tumor cells, leading to a secretion of various factors belonging to a family of damage-associated molecular patterns molecules, capable of inducing the adaptive immune response. One of them is calreticulin (CRT), an endoplasmic reticulum-associated chaperone. Its presence on the surface of dying tumor cells serves as an “eat me” signal for antigen presenting cells (APC). Engulfment of tumor cells by APCs results in the presentation of tumor’s antigens to cytotoxic T-cells and production of cytokines/chemokines, which activate immune cells responsible for tumor cells killing. Thus, the development of ICD and the expression of CRT can help standard therapy to eradicate tumor cells. Here, we review the physiological functions of CRT and its involvement in the ICD appearance in malignant dis- ease. Moreover, we also focus on the ability of various anti-cancer drugs to induce expression of surface CRT on ovarian cancer cells. The second aim of this work is to discuss and summarize the prognostic/predictive value of CRT in ovarian cancer patients. -

Supplemental Table 3 - Male Genes Differentially Expressed > 1.5-Fold Among Strains in E11.5 XY Gonads
Supplemental Table 3 - Male genes differentially expressed > 1.5-fold among strains in E11.5 XY gonads. Male genes differentially expressed between C57BL/6J and 129S1/SvImJ. Note: Positive fold values reflect male genes that are up regulated in C57BL/6J relative to 129S1/SvImJ. Fold Diff Gene symbol Genbank acc Description 10.77 Gcnt1 NM_173442 Mus musculus glucosaminyl (N-acetyl) transferase 1, core 2 (Gcnt1), mRNA [NM_173442] 5.50 Afp NM_007423 Mus musculus alpha fetoprotein (Afp), mRNA [NM_007423] 4.95 Hnf4a NM_008261 Mus musculus hepatic nuclear factor 4, alpha (Hnf4a), mRNA [NM_008261] 4.71 Ppp1r14c AK082372 Mus musculus 0 day neonate cerebellum cDNA, RIKEN full-length enriched library, clone:C230042N14 product:hypothetical protein, full insert sequence. [AK082372] 4.41 Gorasp2 AK020521 Mus musculus 12 days embryo embryonic body between diaphragm region and neck cDNA, RIKEN full-length enriched library, clone:9430094F20 product:inferred: golgi reassembly stacking protein 2, full insert sequence. [AK020521] 3.69 Tmc7 NM_172476 Mus musculus transmembrane channel-like gene family 7 (Tmc7), mRNA [NM_172476] 2.97 Mt2 NM_008630 Mus musculus metallothionein 2 (Mt2), mRNA [NM_008630] 2.62 Gstm6 NM_008184 Mus musculus glutathione S-transferase, mu 6 (Gstm6), mRNA [NM_008184] 2.43 Adhfe1 NM_175236 Mus musculus alcohol dehydrogenase, iron containing, 1 (Adhfe1), mRNA [NM_175236] 2.38 Txndc2 NM_153519 Mus musculus thioredoxin domain containing 2 (spermatozoa) (Txndc2), mRNA [NM_153519] 2.30 C030038J10Rik AK173336 Mus musculus mRNA for mKIAA2027 -
Primepcr™Assay Validation Report
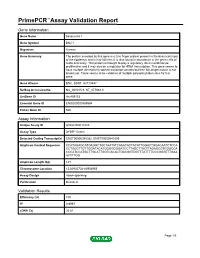
PrimePCR™Assay Validation Report Gene Information Gene Name basonuclin 1 Gene Symbol BNC1 Organism Human Gene Summary The protein encoded by this gene is a zinc finger protein present in the basal cell layer of the epidermis and in hair follicles. It is also found in abundance in the germ cells of testis and ovary. This protein is thought to play a regulatory role in keratinocyte proliferation and it may also be a regulator for rRNA transcription. This gene seems to have multiple alternatively spliced transcript variants but their full-length nature is not known yet. There seems to be evidence of multiple polyadenylation sites for this gene. Gene Aliases BNC, BSN1, HsT19447 RefSeq Accession No. NC_000015.9, NT_077661.3 UniGene ID Hs.459153 Ensembl Gene ID ENSG00000169594 Entrez Gene ID 646 Assay Information Unique Assay ID qHsaCID0017223 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000345382, ENST00000541809 Amplicon Context Sequence CCATAGAGCATGAGGCTGCTAATATCAAACACTACATTGGACTGGACAATCTCCA CCTGGCTTGTTGGATACATGGGGGGGATCCTTAGCTTACTTAGAGCGTGGGCCA CCCATCCATGCTTGCATTGGTCACACTGACGGTGGTTTATTTTCCCGGGTTTGAA ACTTTGG Amplicon Length (bp) 141 Chromosome Location 15:83935724-83936955 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9997 cDNA Cq 26.81 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 84.5 gDNA Cq 39.42 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report BNC1, Human Amplification -

NOD-Like Receptors in the Eye: Uncovering Its Role in Diabetic Retinopathy
International Journal of Molecular Sciences Review NOD-like Receptors in the Eye: Uncovering Its Role in Diabetic Retinopathy Rayne R. Lim 1,2,3, Margaret E. Wieser 1, Rama R. Ganga 4, Veluchamy A. Barathi 5, Rajamani Lakshminarayanan 5 , Rajiv R. Mohan 1,2,3,6, Dean P. Hainsworth 6 and Shyam S. Chaurasia 1,2,3,* 1 Ocular Immunology and Angiogenesis Lab, University of Missouri, Columbia, MO 652011, USA; [email protected] (R.R.L.); [email protected] (M.E.W.); [email protected] (R.R.M.) 2 Department of Biomedical Sciences, University of Missouri, Columbia, MO 652011, USA 3 Ophthalmology, Harry S. Truman Memorial Veterans’ Hospital, Columbia, MO 652011, USA 4 Surgery, University of Missouri, Columbia, MO 652011, USA; [email protected] 5 Singapore Eye Research Institute, Singapore 169856, Singapore; [email protected] (V.A.B.); [email protected] (R.L.) 6 Mason Eye Institute, School of Medicine, University of Missouri, Columbia, MO 652011, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-573-882-3207 Received: 9 December 2019; Accepted: 27 January 2020; Published: 30 January 2020 Abstract: Diabetic retinopathy (DR) is an ocular complication of diabetes mellitus (DM). International Diabetic Federations (IDF) estimates up to 629 million people with DM by the year 2045 worldwide. Nearly 50% of DM patients will show evidence of diabetic-related eye problems. Therapeutic interventions for DR are limited and mostly involve surgical intervention at the late-stages of the disease. The lack of early-stage diagnostic tools and therapies, especially in DR, demands a better understanding of the biological processes involved in the etiology of disease progression. -

Genetic and Genomic Analysis of Hyperlipidemia, Obesity and Diabetes Using (C57BL/6J × TALLYHO/Jngj) F2 Mice
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Nutrition Publications and Other Works Nutrition 12-19-2010 Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P. Stewart Marshall University Hyoung Y. Kim University of Tennessee - Knoxville, [email protected] Arnold M. Saxton University of Tennessee - Knoxville, [email protected] Jung H. Kim Marshall University Follow this and additional works at: https://trace.tennessee.edu/utk_nutrpubs Part of the Animal Sciences Commons, and the Nutrition Commons Recommended Citation BMC Genomics 2010, 11:713 doi:10.1186/1471-2164-11-713 This Article is brought to you for free and open access by the Nutrition at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Nutrition Publications and Other Works by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. Stewart et al. BMC Genomics 2010, 11:713 http://www.biomedcentral.com/1471-2164/11/713 RESEARCH ARTICLE Open Access Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P Stewart1, Hyoung Yon Kim2, Arnold M Saxton3, Jung Han Kim1* Abstract Background: Type 2 diabetes (T2D) is the most common form of diabetes in humans and is closely associated with dyslipidemia and obesity that magnifies the mortality and morbidity related to T2D. The genetic contribution to human T2D and related metabolic disorders is evident, and mostly follows polygenic inheritance. The TALLYHO/ JngJ (TH) mice are a polygenic model for T2D characterized by obesity, hyperinsulinemia, impaired glucose uptake and tolerance, hyperlipidemia, and hyperglycemia. -

Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’S Biological Network Based on Systematic Pharmacology
ORIGINAL RESEARCH published: 25 June 2021 doi: 10.3389/fphar.2021.601846 Exploring the Regulatory Mechanism of Hedysarum Multijugum Maxim.-Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’s Biological Network Based on Systematic Pharmacology Kailin Yang 1†, Liuting Zeng 1†, Anqi Ge 2†, Yi Chen 1†, Shanshan Wang 1†, Xiaofei Zhu 1,3† and Jinwen Ge 1,4* Edited by: 1 Takashi Sato, Key Laboratory of Hunan Province for Integrated Traditional Chinese and Western Medicine on Prevention and Treatment of 2 Tokyo University of Pharmacy and Life Cardio-Cerebral Diseases, Hunan University of Chinese Medicine, Changsha, China, Galactophore Department, The First 3 Sciences, Japan Hospital of Hunan University of Chinese Medicine, Changsha, China, School of Graduate, Central South University, Changsha, China, 4Shaoyang University, Shaoyang, China Reviewed by: Hui Zhao, Capital Medical University, China Background: Clinical research found that Hedysarum Multijugum Maxim.-Chuanxiong Maria Luisa Del Moral, fi University of Jaén, Spain Rhizoma Compound (HCC) has de nite curative effect on cerebral ischemic diseases, *Correspondence: such as ischemic stroke and cerebral ischemia-reperfusion injury (CIR). However, its Jinwen Ge mechanism for treating cerebral ischemia is still not fully explained. [email protected] †These authors share first authorship Methods: The traditional Chinese medicine related database were utilized to obtain the components of HCC. The Pharmmapper were used to predict HCC’s potential targets. Specialty section: The CIR genes were obtained from Genecards and OMIM and the protein-protein This article was submitted to interaction (PPI) data of HCC’s targets and IS genes were obtained from String Ethnopharmacology, a section of the journal database. -

ATP-Binding and Hydrolysis in Inflammasome Activation
molecules Review ATP-Binding and Hydrolysis in Inflammasome Activation Christina F. Sandall, Bjoern K. Ziehr and Justin A. MacDonald * Department of Biochemistry & Molecular Biology, Cumming School of Medicine, University of Calgary, 3280 Hospital Drive NW, Calgary, AB T2N 4Z6, Canada; [email protected] (C.F.S.); [email protected] (B.K.Z.) * Correspondence: [email protected]; Tel.: +1-403-210-8433 Academic Editor: Massimo Bertinaria Received: 15 September 2020; Accepted: 3 October 2020; Published: 7 October 2020 Abstract: The prototypical model for NOD-like receptor (NLR) inflammasome assembly includes nucleotide-dependent activation of the NLR downstream of pathogen- or danger-associated molecular pattern (PAMP or DAMP) recognition, followed by nucleation of hetero-oligomeric platforms that lie upstream of inflammatory responses associated with innate immunity. As members of the STAND ATPases, the NLRs are generally thought to share a similar model of ATP-dependent activation and effect. However, recent observations have challenged this paradigm to reveal novel and complex biochemical processes to discern NLRs from other STAND proteins. In this review, we highlight past findings that identify the regulatory importance of conserved ATP-binding and hydrolysis motifs within the nucleotide-binding NACHT domain of NLRs and explore recent breakthroughs that generate connections between NLR protein structure and function. Indeed, newly deposited NLR structures for NLRC4 and NLRP3 have provided unique perspectives on the ATP-dependency of inflammasome activation. Novel molecular dynamic simulations of NLRP3 examined the active site of ADP- and ATP-bound models. The findings support distinctions in nucleotide-binding domain topology with occupancy of ATP or ADP that are in turn disseminated on to the global protein structure. -

The DNA Methylation Landscape of Glioblastoma Disease Progression Shows Extensive Heterogeneity in Time and Space
bioRxiv preprint doi: https://doi.org/10.1101/173864; this version posted August 9, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space Johanna Klughammer1*, Barbara Kiesel2,3*, Thomas Roetzer3,4, Nikolaus Fortelny1, Amelie Kuchler1, Nathan C. Sheffield5, Paul Datlinger1, Nadine Peter3,4, Karl-Heinz Nenning6, Julia Furtner3,7, Martha Nowosielski8,9, Marco Augustin10, Mario Mischkulnig2,3, Thomas Ströbel3,4, Patrizia Moser11, Christian F. Freyschlag12, Jo- hannes Kerschbaumer12, Claudius Thomé12, Astrid E. Grams13, Günther Stockhammer8, Melitta Kitzwoegerer14, Stefan Oberndorfer15, Franz Marhold16, Serge Weis17, Johannes Trenkler18, Johanna Buchroithner19, Josef Pichler20, Johannes Haybaeck21,22, Stefanie Krassnig21, Kariem Madhy Ali23, Gord von Campe23, Franz Payer24, Camillo Sherif25, Julius Preiser26, Thomas Hauser27, Peter A. Winkler27, Waltraud Kleindienst28, Franz Würtz29, Tanisa Brandner-Kokalj29, Martin Stultschnig30, Stefan Schweiger31, Karin Dieckmann3,32, Matthias Preusser3,33, Georg Langs6, Bernhard Baumann10, Engelbert Knosp2,3, Georg Widhalm2,3, Christine Marosi3,33, Johannes A. Hainfellner3,4, Adelheid Woehrer3,4#§, Christoph Bock1,34,35# 1 CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria. 2 Department of Neurosurgery, Medical University of Vienna, Vienna, Austria. 3 Comprehensive Cancer Center, Central Nervous System Tumor Unit, Medical University of Vienna, Austria. 4 Institute of Neurology, Medical University of Vienna, Vienna, Austria. 5 Center for Public Health Genomics, University of Virginia, Charlottesville VA, USA. 6 Department of Biomedical Imaging and Image-guided Therapy, Computational Imaging Research Lab, Medical University of Vi- enna, Vienna, Austria. -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas.