Thermodynamic Concepts and Processes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
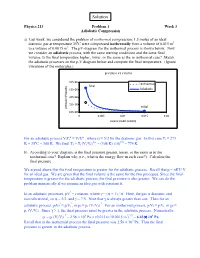
3-1 Adiabatic Compression
Solution Physics 213 Problem 1 Week 3 Adiabatic Compression a) Last week, we considered the problem of isothermal compression: 1.5 moles of an ideal diatomic gas at temperature 35oC were compressed isothermally from a volume of 0.015 m3 to a volume of 0.0015 m3. The pV-diagram for the isothermal process is shown below. Now we consider an adiabatic process, with the same starting conditions and the same final volume. Is the final temperature higher, lower, or the same as the in isothermal case? Sketch the adiabatic processes on the p-V diagram below and compute the final temperature. (Ignore vibrations of the molecules.) α α For an adiabatic process ViTi = VfTf , where α = 5/2 for the diatomic gas. In this case Ti = 273 o 1/α 2/5 K + 35 C = 308 K. We find Tf = Ti (Vi/Vf) = (308 K) (10) = 774 K. b) According to your diagram, is the final pressure greater, lesser, or the same as in the isothermal case? Explain why (i.e., what is the energy flow in each case?). Calculate the final pressure. We argued above that the final temperature is greater for the adiabatic process. Recall that p = nRT/ V for an ideal gas. We are given that the final volume is the same for the two processes. Since the final temperature is greater for the adiabatic process, the final pressure is also greater. We can do the problem numerically if we assume an idea gas with constant α. γ In an adiabatic processes, pV = constant, where γ = (α + 1) / α. -

Recitation: 3 9/18/03
Recitation: 3 9/18/03 Second Law ² There exists a function (S) of the extensive parameters of any composite system, defined for all equilibrium states and having the following property: The values assumed by the extensive parameters in the absence of an internal constraint are those that maximize the entropy over the manifold of constrained equilibrium states. ¡¢ ¡ ² Another way of looking at it: ¡¢ ¡ U1 U2 U=U1+U2 S1 S2 S>S1+S2 ¡¢ ¡ Equilibrium State Figure 1: Second Law ² Entropy is additive. And... � ¶ @S > 0 @U V;N::: ² S is an extensive property: It is a homogeneous, first order function of extensive parameters: S (¸U; ¸V; ¸N) =¸S (U; V; N) ² S is not conserved. ±Q dSSys ¸ TSys For an Isolated System, (i.e. Universe) 4S > 0 Locally, the entropy of the system can decrease. However, this must be compensated by a total increase in the entropy of the universe. (See 2) Q Q ΔS1=¡ ΔS2= + T1³ ´ T2 1 1 ΔST otal =Q ¡ > 0 T2 T1 QuasiStatic Processes A Quasistatic thermodynamic process is defined as the trajectory, in thermodynamic space, along an infinite number of contiguous equilibrium states that connect to equilibrium states, A and B. Universe System T2 T1 Q T1>T2 Figure 2: Local vs. Total Change in Energy Irreversible Process Consider a closed system that can go from state A to state B. The system is induced to go from A to B through the removal of some internal constraint (e.g. removal of adiabatic wall). The systems moves to state B only if B has a maximum entropy with respect to all the other accessible states. -

Guggenheim Aeronautical Laboratory~://; California
R.ES~.LttCH REPCRT GUGGENHEIM AERONAUTICAL LABORATORY ~://; CALIFORNIA INSTITUTE OF TECHNOLOGY HYPERSC»JIC RESEARCH PROJECT Memorandum No. 47 December 15, 1958 AN EXPERIMENTAL INVESTIGATION OF THE EFFECT OF EJECTING A COOLANT GAS AT THE NOSE OF A BLUNT BODY by C. Hugh E. Warren ARMY ORDNANCE CONTRACT NO. DA-04-495-0rd-19 GUGGENHEIM AERONAUTICAL LABORATORY CALIFORNIA INSTITUTE OF TECHNOLOGY Pasadena1 California HYPERSONIC RESEARCH PROJECT Memorandum No. 4 7 December 151 1958 AN EXPERIMENTAL INVESTIGATION OF THE EFFECT OF EJECTING A COOLANT GAS AT THE NOSE OF A BLUNT BODY by C. Hugh E. Warren ~~C!a r~:Ml ia:nl bil'eCtOr Guggenheim Aeronautical Laboratory ARMY ORDNANCE CONTRACT NO. DA-04-495-0rd-19 Army Project No. 5B0306004 Ordnance Project No. TB3-0118 OOR Project No. 1600-PE ACKNOWLEDGMENTS The author wishes to express his appreciation to Professor Lester Lees for his assistance1 guidance and encouragement during this work1 to Mr. Howard McDonald and other members of the Aeronautics Machine Shop for fabricating and maintaining the models and other items of equipment1 to Mr. Paul Baloga and the staff of the Hypersonic Wind Tunnel for their assistance and advice during testing~ to Mrs. Betty Laue for much computational work1 and to Mrs. Gerry Van Gieson for typing the manuscript and piloting the report through after the author 1 s departure. Most of the work was done while the author was in receipt of a Harkness Fellowship of the Commonwealth Fund of New York1 to whom thanks are due for the opportunity to spend a year on such an instructive and interesting program in such congenial surroundings. -

Thermodynamics Formulation of Economics Burin Gumjudpai A
Thermodynamics Formulation of Economics 306706052 NIDA E-THESIS 5710313001 thesis / recv: 09012563 15:07:41 seq: 16 Burin Gumjudpai A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Economics (Financial Economics) School of Development Economics National Institute of Development Administration 2019 Thermodynamics Formulation of Economics Burin Gumjudpai School of Development Economics Major Advisor (Associate Professor Yuthana Sethapramote, Ph.D.) The Examining Committee Approved This Thesis Submitted in Partial 306706052 Fulfillment of the Requirements for the Degree of Master of Economics (Financial Economics). Committee Chairperson NIDA E-THESIS 5710313001 thesis / recv: 09012563 15:07:41 seq: 16 (Assistant Professor Pongsak Luangaram, Ph.D.) Committee (Assistant Professor Athakrit Thepmongkol, Ph.D.) Committee (Associate Professor Yuthana Sethapramote, Ph.D.) Dean (Associate Professor Amornrat Apinunmahakul, Ph.D.) ______/______/______ ABST RACT ABSTRACT Title of Thesis Thermodynamics Formulation of Economics Author Burin Gumjudpai Degree Master of Economics (Financial Economics) Year 2019 306706052 We consider a group of information-symmetric consumers with one type of commodity in an efficient market. The commodity is fixed asset and is non-disposable. We are interested in seeing if there is some connection of thermodynamics formulation NIDA E-THESIS 5710313001 thesis / recv: 09012563 15:07:41 seq: 16 to microeconomics. We follow Carathéodory approach which requires empirical existence of equation of state (EoS) and coordinates before performing maximization of variables. In investigating EoS of various system for constructing the economics EoS, unexpectedly new insights of thermodynamics are discovered. With definition of truly endogenous function, criteria rules and diagrams are proposed in identifying the status of EoS for an empirical equation. -

Chapter 7 BOUNDARY WORK
Chapter 7 BOUNDARY WORK Frank is struck - as if for the first time - by how much civilization depends on not seeing certain things and pretending others never occurred. − Francine Prose (Amateur Voodoo) In this chapter we will learn to evaluate Win, which is the work term in the first law of thermodynamics. Work can be of different types, such as boundary work, stirring work, electrical work, magnetic work, work of changing the surface area, and work to overcome friction. In this chapter, we will concentrate on the evaluation of boundary work. 110 Chapter 7 7.1 Boundary Work in Real Life Boundary work is done when the boundary of the system moves, causing either compression or expansion of the system. A real-life application of boundary work, for example, is found in the diesel engine which consists of pistons and cylinders as the prime component of the engine. In a diesel engine, air is fed to the cylinder and is compressed by the upward movement of the piston. The boundary work done by the piston in compressing the air is responsible for the increase in air temperature. Onto the hot air, diesel fuel is sprayed, which leads to spontaneous self ignition of the fuel-air mixture. The chemical energy released during this combustion process would heat up the gases produced during combustion. As the gases expand due to heating, they would push the piston downwards with great force. A major portion of the boundary work done by the gases on the piston gets converted into the energy required to rotate the shaft of the engine. -

Application of Laser Doppler Velocimetry
APPLICATION OF LASER DOPPLER VELOCIMETRY (LDV) TO STUDY THE STRUCTURE OF GRAVITY CURRENTS UNDER FIRE CONDITIONS B Moghtaderi† Process Safety and Environment Protection Group Discipline of Chemical Engineering, School of Engineering Faculty of Engineering & Built Environment, The University of Newcastle Callaghan, NSW 2308, Australia ABSTRACT Gravity currents are of considerable safety importance primarily because of their role in the spread and transport of smoke and hot gases in building fires. Despite recent progress in the field, relatively little is known about the structure of gravity currents under conditions pertinent to building fires. The present investigation is an attempt to address this shortcoming by studying the turbulent structure of gravity currents. For this purpose, a series of experiments was conducted in a rectangular tank with turbulent, sub-critical underflows. Laser-Doppler Velocimetry was employed to quantify the velocity field and associated turbulent flow parameters. Experimental results indicated that the mean flow within the head region primarily consisted of an undiluted large single vortex which rapidly mixed with the ambient flow in the wake region. Cases with isothermal wall boundary conditions showed three-dimensional effects whereas those with adiabatic walls exhibited two-dimensional behaviour. Turbulence was found to be highly heterogeneous and its distribution was governed by the location of large eddies. While all components of turbulence kinetic energy showed minima in the regions where velocity was maximum (i.e. low fluid shear), they reached their maximum in the shear layer at the upper boundary of the flow. KEY WORDS: gravity currents, laser Doppler velocimetry, turbulent structure INTRODUCTION Gravity currents are important physical phenomena which have direct implications in a wide variety of physical situations ranging from environmental phenomena‡ to enclosure fires. -

Conduction Heat Transfer Notes for MECH 7210
Conduction Heat Transfer Notes for MECH 7210 Daniel W. Mackowski Mechanical Engineering Department Auburn University 2 Preface The Notes on Conduction Heat Transfer are, as the name suggests, a compilation of lecture notes put together over 10 years of teaching the subject. The notes are not meant to be a comprehensive ∼ presentation of the subject of heat conduction, and the student is referred to the texts referenced below for such treatments. A goal of mine, in preparing the notes, has been to address an apparent shortcoming in many of the current texts, in that the texts present the mathematical formulation and analytical solution to a wide variety of conduction problems, yet they spend little if any time on discussing how numerical and graphical results can be obtained from the solutions. As will be seen, this task in itself is not trivial, and to this end mathematical software packages (in particular, the package Mathematica) will be used extensively in application of the analytical solutions. The notes were prepared using the LATEX typesetting program, which is freely available via internet download. I wish to thank my former students, who have (and continue) to catch the multitude of mistakes and typos in the notes. These notes are dedicated to the memory of Clifford Cremers, an outstanding teacher of heat transfer and a fine fly fisherman. References The ‘text’ in the course will consist of my lecture notes – which contain few if any literature citations. I will need to fix this if I ever expect to publish the notes as a book. The following reference texts were used to prepare the notes. -

The First Law of Thermodynamics Continued Pre-Reading: §19.5 Where We Are
Lecture 7 The first law of thermodynamics continued Pre-reading: §19.5 Where we are The pressure p, volume V, and temperature T are related by an equation of state. For an ideal gas, pV = nRT = NkT For an ideal gas, the temperature T is is a direct measure of the average kinetic energy of its 3 3 molecules: KE = nRT = NkT tr 2 2 2 3kT 3RT and vrms = (v )av = = r m r M p Where we are We define the internal energy of a system: UKEPE=+∑∑ interaction Random chaotic between atoms motion & molecules For an ideal gas, f UNkT= 2 i.e. the internal energy depends only on its temperature Where we are By considering adding heat to a fixed volume of an ideal gas, we showed f f Q = Nk∆T = nR∆T 2 2 and so, from the definition of heat capacity Q = nC∆T f we have that C = R for any ideal gas. V 2 Change in internal energy: ∆U = nCV ∆T Heat capacity of an ideal gas Now consider adding heat to an ideal gas at constant pressure. By definition, Q = nCp∆T and W = p∆V = nR∆T So from ∆U = Q W − we get nCV ∆T = nCp∆T nR∆T − or Cp = CV + R It takes greater heat input to raise the temperature of a gas a given amount at constant pressure than constant volume YF §19.4 Ratio of heat capacities Look at the ratio of these heat capacities: we have f C = R V 2 and f + 2 C = C + R = R p V 2 so C p γ = > 1 CV 3 For a monatomic gas, CV = R 3 5 2 so Cp = R + R = R 2 2 C 5 R 5 and γ = p = 2 = =1.67 C 3 R 3 YF §19.4 V 2 Problem An ideal gas is enclosed in a cylinder which has a movable piston. -

Adiabatic Bulk Moduli
8.03 at ESG Supplemental Notes Adiabatic Bulk Moduli To find the speed of sound in a gas, or any property of a gas involving elasticity (see the discussion of the Helmholtz oscillator, B&B page 22, or B&B problem 1.6, or French Pages 57-59.), we need the “bulk modulus” of the fluid. This will correspond to the “spring constant” of a spring, and will give the magnitude of the restoring agency (pressure for a gas, force for a spring) in terms of the change in physical dimension (volume for a gas, length for a spring). It turns out to be more useful to use an intensive quantity for the bulk modulus of a gas, so what we want is the change in pressure per fractional change in volume, so the bulk modulus, denoted as κ (the Greek “kappa” which, when written, has a great tendency to look like k, and in fact French uses “K”), is ∆p dp κ = − , or κ = −V . ∆V/V dV The minus sign indicates that for normal fluids (not all are!), a negative change in volume results in an increase in pressure. To find the bulk modulus, we need to know something about the gas and how it behaves. For our purposes, we will need three basic principles (actually 2 1/2) which we get from thermodynamics, or the kinetic theory of gasses. You might well have encountered these in previous classes, such as chemistry. A) The ideal gas law; pV = nRT , with the standard terminology. B) The first law of thermodynamics; dU = dQ − pdV,whereUis internal energy and Q is heat. -

Session 15 Thermodynamics
Session 15 Thermodynamics Cheryl Hurkett Physics Innovations Centre for Excellence in Learning and Teaching CETL (Leicester) Department of Physics and Astronomy University of Leicester 1 Contents Welcome .................................................................................................................................. 4 Session Authors .................................................................................................................. 4 Learning Objectives .............................................................................................................. 5 The Problem ........................................................................................................................... 6 Issues .................................................................................................................................... 6 Energy, Heat and Work ........................................................................................................ 7 The First Law of thermodynamics................................................................................... 8 Work..................................................................................................................................... 9 Heat .................................................................................................................................... 11 Principal Specific heats of a gas ..................................................................................... 12 Summary .......................................................................................................................... -

Thermodynamic Processes: the Limits of Possible
Thermodynamic Processes: The Limits of Possible Thermodynamics put severe restrictions on what processes involving a change of the thermodynamic state of a system (U,V,N,…) are possible. In a quasi-static process system moves from one equilibrium state to another via a series of other equilibrium states . All quasi-static processes fall into two main classes: reversible and irreversible processes . Processes with decreasing total entropy of a thermodynamic system and its environment are prohibited by Postulate II Notes Graphic representation of a single thermodynamic system Phase space of extensive coordinates The fundamental relation S(1) =S(U (1) , X (1) ) of a thermodynamic system defines a hypersurface in the coordinate space S(1) S(1) U(1) U(1) X(1) X(1) S(1) – entropy of system 1 (1) (1) (1) (1) (1) U – energy of system 1 X = V , N 1 , …N m – coordinates of system 1 Notes Graphic representation of a composite thermodynamic system Phase space of extensive coordinates The fundamental relation of a composite thermodynamic system S = S (1) (U (1 ), X (1) ) + S (2) (U-U(1) ,X (2) ) (system 1 and system 2). defines a hyper-surface in the coordinate space of the composite system S(1+2) S(1+2) U (1,2) X = V, N 1, …N m – coordinates U of subsystems (1 and 2) X(1,2) (1,2) S – entropy of a composite system X U – energy of a composite system Notes Irreversible and reversible processes If we change constraints on some of the composite system coordinates, (e.g. -

1 Microphysics
ASTR 501 Stellar Physics 1 1 Microphysics • equation of state, density: ρ(P, T, X) • radiative absorption coefficient, opacity: κ(P, T, X) • rate of energy production: (P, T, X) (nuclear physics) Nuclear physics also responsible for composition changes dXj = Fˆj · Xj (1) dt burn while the total composition change also includes a mixing term: dX ∂ dX i = 4πr2ρ D i (2) dt mix ∂m dm 1.1 Equation of state For the fluid to have an EOS it must be collisional: lregion λ (3) where λ is the mean free path. P from equation of state, consider three cases: ASTR 501 Stellar Physics 2 (a) ideal gas equation of state: p = R ρT (b) barotropic equation of state: pressure p depends only on ρ, as for example • isothermal: p ∼ ρ • adiabatic: p = Kργ Plus non-ideal effects, e.g. electron degeneracy. 1.2 Ideal gas equation of state Assumption: internal energy entirely in kinetic energy according to kinetic theory: = CVT , where CV is the specific heat at constant volume. This can be seen from the first law of thermodynamics dQ = d + pdV , where V is the specific volume, d if we express d = dT dT . d Similarly, the specific heat at constant pressure Cp = dT + R where R is the gas constant. This follows from the ideal gas equation of state. [†13] It also follows that Cp − CV = R [†14], and the ratio of specific heats is defined as C γ = p . (4) CV ASTR 501 Stellar Physics 3 α CV can be related to R as CV = 2 R , where α is the number of degrees of freedom 1 per particle, since the energy per particle per degree of freedom is 2kT where k is 2+α the Boltzmann constant, and R = nk.