Pathogenesis of Prion Diseases: a Progress Report
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Epidemiology Bulletin
VIRGINIA EPIDEMIOLOGY BULLETIN Robert B. Stroube, M.D., M.P.H., Health Commissioner Christopher Novak, M.D., M.P.H., Editor Suzanne R. Jenkins, V.M.D., M.P.H., Acting State Epidemiologist Vickie L. O’Dell, Layout Editor January 2005 Volume 105, No. 1 Transmissible Spongiform Encephalopathies (TSEs) A little over one year ago, on Decem- of diseases known as transmissible ber 25, 2003, the US Department of Agri- spongiform encephalopathies (TSEs). This a b culture (USDA) confirmed the diagnosis article is part of the Virginia Department of bovine spongiform encephalopathy of Health’s efforts to improve surveillance (BSE, or “mad cow disease”) in a single for TSEs in Virginia to better understand 1 dairy cow in Washington state. As a re- the disease as well as to detect any conversion sult, the USDA expanded and intensified changes in disease patterns. its national testing program for BSE. From June 1, 2004, to January 2, 2005, 167,476 Overview of TSEs alpha-helices beta-sheets cattle were tested. Not a single positive TSEs are a group of relatively rare pro- 2 animal was found. gressive neurodegenerative disorders that In May 2004, the New Jersey Depart- affect both humans and animals. They are Figure 1. (a) A healthy cellular ment of Health and Senior Services distinguished by long incubation periods, prion protein contains only alpha- (NJDHSS) and the Centers for Disease characteristic spongiform changes of the helices. (b) The pathogenic isoform Control and Prevention (CDC) reported brain associated with neuronal loss, and with alpha-helical structures on a suspected cluster of Creutzfeldt- the lack of a detectable immune or inflam- converted to beta-sheets.6 Jakob disease (CJD) cases (including one matory response. -

Vaccination with Prion Peptide-Displaying Polyomavirus-Like Particles Prolongs Incubation Time in Scrapie-Infected Mice
viruses Article Vaccination with Prion Peptide-Displaying Polyomavirus-Like Particles Prolongs Incubation Time in Scrapie-Infected Mice Martin Eiden 1,* , Alma Gedvilaite 2 , Fabienne Leidel 1,3, Rainer G. Ulrich 1 and Martin H. Groschup 1 1 Institute of Novel and Emerging Infectious Diseases, Friedrich-Loeffler-Institut, Federal Research Institute for Animal Health, Südufer 10, 17493 Greifswald-Insel Riems, Germany; [email protected] (F.L.); rainer.ulrich@fli.de (R.G.U.); martin.groschup@fli.de (M.H.G.) 2 Life Sciences Center, Institute of Biotechnology, Vilnius University, Sauletekio˙ al. 7, LT-10257 Vilnius, Lithuania; [email protected] 3 Task Force Animal Diseases, Darmstadt Regional Administrative Council, Luisenplatz 2, 64283 Darmstadt, Germany * Correspondence: martin.eiden@fli.de Abstract: Prion diseases like scrapie in sheep, bovine spongiform encephalopathy (BSE) in cattle or Creutzfeldt–Jakob disease (CJD) in humans are fatal neurodegenerative diseases characterized by the conformational conversion of the normal, mainly α-helical cellular prion protein (PrPC) into the abnormal β-sheet rich infectious isoform PrPSc. Various therapeutic or prophylactic approaches have been conducted, but no approved therapeutic treatment is available so far. Immunisation against prions is hampered by the self-tolerance to PrPC in mammalian species. One strategy to avoid this tolerance is presenting PrP variants in virus-like particles (VLPs). Therefore, we vaccinated C57/BL6 mice with nine prion peptide variants presented by hamster polyomavirus capsid protein VP1/VP2-derived VLPs. Mice were subsequently challenged intraperitoneally with the murine RML Citation: Eiden, M.; Gedvilaite, A.; prion strain. Importantly, one group exhibited significantly increased mean survival time of 240 days Leidel, F.; Ulrich, R.G.; Groschup, M.H. -

Scrapie and Tagging/Tattooing Your Goats and Sheep 2017 PRESENTATION for ASHTABULA COUNTY 4-H by STEPHANIE MAROUS What Is Scrapie?
Scrapie and Tagging/Tattooing Your Goats and Sheep 2017 PRESENTATION FOR ASHTABULA COUNTY 4-H BY STEPHANIE MAROUS What is Scrapie? Scrapie is a fatal degenerative disease of the central nervous system of sheep and goats. (Basically it is the sheep and goat version of Mad Cow Disease) Scrapie is commonly spread from a female to her offspring. Other members of the herd can catch it through contact with the placenta or its fluids. Scrapie can only truly be tested after an animal is dead. This is done by testing the brain tissue and looking for the disease. SYMPTOMS Symptoms may take 2-5 years to appear Head and Neck Tremors Skin Itching (this is where the term scrapie comes from) Inability to control legs (remember this attacks the Nervous System) Good appetite accompanied by weight loss. Remember just because an animal exhibits these symptoms does not mean it has scrapie. Consult your Vet to rule out possible reasons for symptoms. Scrapie Eradication Program The National Scrapie Eradication Program, coordinated by the U.S. Department of Agriculture’s (USDA) Animal and Plant Health Inspection Service (APHIS), has reduced the prevalence of scrapie by over 85 percent. To find and eliminate the last few cases in the United States, the cooperation of sheep and goat producers throughout the country is needed. Producers are required to follow Federal and State regulations for officially identifying their sheep and goats. Producers must also keep herd records showing what new animals were added and what animals left the herd/flock Scrapie Eradication Program APHIS provides official plastic or metal eartags free of charge to producers. -

Zinc and Copper Ions Differentially Regulate Prion-Like Phase
viruses Article Zinc and Copper Ions Differentially Regulate Prion-Like Phase Separation Dynamics of Pan-Virus Nucleocapsid Biomolecular Condensates Anne Monette 1,* and Andrew J. Mouland 1,2,* 1 Lady Davis Institute at the Jewish General Hospital, Montréal, QC H3T 1E2, Canada 2 Department of Medicine, McGill University, Montréal, QC H4A 3J1, Canada * Correspondence: [email protected] (A.M.); [email protected] (A.J.M.) Received: 2 September 2020; Accepted: 12 October 2020; Published: 18 October 2020 Abstract: Liquid-liquid phase separation (LLPS) is a rapidly growing research focus due to numerous demonstrations that many cellular proteins phase-separate to form biomolecular condensates (BMCs) that nucleate membraneless organelles (MLOs). A growing repertoire of mechanisms supporting BMC formation, composition, dynamics, and functions are becoming elucidated. BMCs are now appreciated as required for several steps of gene regulation, while their deregulation promotes pathological aggregates, such as stress granules (SGs) and insoluble irreversible plaques that are hallmarks of neurodegenerative diseases. Treatment of BMC-related diseases will greatly benefit from identification of therapeutics preventing pathological aggregates while sparing BMCs required for cellular functions. Numerous viruses that block SG assembly also utilize or engineer BMCs for their replication. While BMC formation first depends on prion-like disordered protein domains (PrLDs), metal ion-controlled RNA-binding domains (RBDs) also orchestrate their formation. Virus replication and viral genomic RNA (vRNA) packaging dynamics involving nucleocapsid (NC) proteins and their orthologs rely on Zinc (Zn) availability, while virus morphology and infectivity are negatively influenced by excess Copper (Cu). While virus infections modify physiological metal homeostasis towards an increased copper to zinc ratio (Cu/Zn), how and why they do this remains elusive. -

Chronic Wasting Disease and Atypical Forms of Bovine Spongiform
Journal of General Virology (2012), 93, 1624–1629 DOI 10.1099/vir.0.042507-0 Short Chronic wasting disease and atypical forms of Communication bovine spongiform encephalopathy and scrapie are not transmissible to mice expressing wild-type levels of human prion protein Rona Wilson,1 Chris Plinston,1 Nora Hunter,1 Cristina Casalone,2 Cristiano Corona,2 Fabrizio Tagliavini,3 Silvia Suardi,3 Margherita Ruggerone,3 Fabio Moda,3 Silvia Graziano,4 Marco Sbriccoli,4 Franco Cardone,4 Maurizio Pocchiari,4 Loredana Ingrosso,4 Thierry Baron,5 Juergen Richt,63 Olivier Andreoletti,7 Marion Simmons,8 Richard Lockey,8 Jean C. Manson1 and Rona M. Barron1 Correspondence 1Neuropathogenesis Division, The Roslin Institute and R(D)SVS, University of Edinburgh, Roslin, Rona M. Barron Midlothian, UK [email protected] 2Istituto Zooprofilattico Sperimentale del Piemonte, Liguria e Valle d’Aosta, Turin, Italy 3IRCCS Foundation, ‘Carlo Besta’ Neurological Institute, Milan, Italy 4Department of Cell Biology and Neurosciences, Istituto Superiore di Sanita`, Viale Regina Elena 299, 00161 Rome, Italy 5Agence Nationale de Se´curite´ Sanitaire, Lyon, France 6USDA, ARS, National Animal Disease Center, PO Box 70, Ames, IA 50010, USA 7UMR 1225 Interactions Hoˆtes-Agents Pathoge`nes, INRA, Ecole Nationale Ve´te´rinaire, 23 chemin des Capelles, B.P. 87614, 31076 Toulouse Cedex 3, France 8Neuropathology Section, Department of Pathology and Host Susceptibility, Animal Health and Veterinary Laboratories Agency, Addlestone, Surrey KT15 3NB, UK The association between bovine spongiform encephalopathy (BSE) and variant Creutzfeldt–Jakob disease (vCJD) has demonstrated that cattle transmissible spongiform encephalopathies (TSEs) can pose a risk to human health and raises the possibility that other ruminant TSEs may be transmissible to humans. -

Prion Diseases in Knock-In Mice Carrying Single Prp Codon Substitutions Associated with Human Diseases
Profoundly different prion diseases in knock-in mice carrying single PrP codon substitutions associated with human diseases Walker S. Jacksona,b,c,1, Andrew W. Borkowskia,b,c, Nicki E. Watsona, Oliver D. Kingd, Henryk Faase, Alan Jasanoffe,f, and Susan Lindquista,b,c,2 aWhitehead Institute for Biomedical Research, Cambridge, MA 02142; bHoward Hughes Medical Institute, cDepartment of Biology, and fDepartments of Biological Engineering, Brain and Cognitive Sciences, and Nuclear Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139; dDepartment of Cell and Developmental Biology, University of Massachusetts Medical School, Worcester, MA 01655; and eFrances Bitter Magnet Laboratory, Cambridge, MA 02139 Contributed by Susan Lindquist, July 9, 2013 (sent for review March 7, 2013) In man, mutations in different regions of the prion protein (PrP) linked to it, provides an important general model for such are associated with infectious neurodegenerative diseases that investigations. have remarkably different clinical signs and neuropathological There are several types of human prion diseases, each begin- lesions. To explore the roots of this phenomenon, we created ning with pathologic processes in a different brain region and fi – a knock-in mouse model carrying the mutation associated with leading to distinct functional de cits: cognition [Creutzfeldt – – one of these diseases [Creutzfeldt–Jakob disease (CJD)] that was Jakob disease (CJD)], movement control (Gerstmann Sträussler Scheinker syndrome), or sleep and autonomic functions [fatal exactly analogous to a previous knock-in model of a different fl prion disease [fatal familial insomnia (FFI)]. Together with the familial insomnia (FFI)] (7). Prion diseases also af ict animals WT parent, this created an allelic series of three lines, each express- and include bovine spongiform encephalopathy (BSE) of cattle, scrapie of sheep and goats, and chronic wasting disease (CWD) ing the same protein with a single amino acid difference, and with of deer and elk (1). -

Mitochondrial Dysfunction in Preclinical Genetic Prion Disease: a Target for 7 Preventive Treatment?
1HXURELRORJ\RI'LVHDVH ² Contents lists available at ScienceDirect Neurobiology of Disease journal homepage: www.elsevier.com/locate/ynbdi Mitochondrial dysfunction in preclinical genetic prion disease: A target for 7 preventive treatment? Guy Kellera,c,1, Orli Binyamina,c,1, Kati Frida,c, Ann Saadab,c, Ruth Gabizona,c,⁎ a Department of Neurology, The Agnes Ginges Center for Human Neurogenetics, Israel b Department of Genetics and Metabolic Diseases, The Monique and Jacques Roboh Department of Genetic Research, Hadassah-Hebrew University Medical Center, Israel c Medical School, The Hebrew University, Jerusalem, Israel ABSTRACT Mitochondrial malfunction is a common feature in advanced stages of neurodegenerative conditions, as is the case for the accumulation of aberrantly folded proteins, such as PrP in prion diseases. In this work, we investigated mitochondrial activity and expression of related factors vis a vis PrP accumulation at the subclinical stages of TgMHu2ME199K mice, modeling for genetic prion diseases. While these mice remain healthy until 5–6 months of age, they succumb to fatal disease at 12–14 months. We found that mitochondrial respiratory chain enzymatic activates and ATP/ROS production, were abnormally elevated in asymptomatic mice, concomitant with initial accumulation of disease related PrP. In parallel, the expression of Cytochrome c oxidase (COX) subunit IV isoform 1(Cox IV-1) was reduced and replaced by the activity of Cox IV isoform 2, which operates in oxidative neuronal conditions. At all stages of disease, Cox IV-1 was absent from cells accu- mulating disease related PrP, suggesting that PrP aggregates may directly compromise normal mitochondrial function. Administration of Nano-PSO, a brain targeted antioxidant, to TgMHu2ME199K mice, reversed functional and biochemical mitochondrial functions to normal conditions regardless of the presence of misfolded PrP. -

An Evolutionary Basis for Scrapie Disease : Identification of a Fish
72 Update TRENDS in Genetics Vol.19 No.2 February 2003 affect regulation of the human genome in a more global 11 Frith, M.C. et al. (2002) Statistical significance of clusters of motifs manner by creating S/MARs that form chromatin loops, represented by position specific scoring matrices in nucleotide and by shaping the sequence evolution of LCRs. sequences. Nucleic Acids Res. 30, 3214–3224 12 Wingender, E. et al. (2001) The TRANSFAC system on gene expression regulation. Nucleic Acids Res. 29, 281–283 Acknowledgements 13 Purucker, M. et al. (1990) Structure and function of the enhancer 30 to Galina V. Glazko was supported by research grants from NIH (GM-20293) the human A gamma globin gene. Nucleic Acids Res. 18, 7407–7415 and NASA (NCC2-1057) awarded to Masatoshi Nei. We thank Nathan 14 Li, Q. et al. (1999) Locus control regions: coming of age at a decade plus. J. Bowen and Wolfgang J. Miller for discussions on the relationship Trends Genet. 15, 403–408 between TEs and S/MARs. 15 Hardison, R. et al. (1997) Locus control regions of mammalian beta- globin gene clusters: combining phylogenetic analyses and exper- References imental results to gain functional insights. Gene 205, 73–94 1 The Human Genome Sequencing Consortium, (2001) Initial sequen- 16 Strausberg, R. et al. (1999) The mammalian gene collection. Science cing and analysis of the human genome. Nature 409, 860–921 286, 455–457 2 Kidwell, M.G. and Lisch, D.R. (2001) Perspective: transposable 17 Bode, J. et al. (1996) Scaffold/matrix-attached regions: topological elements, parasitic DNA, and genome evolution. -

An Overview of the Disease and the National Scrapie Eradication Program
Scrapie An overview of the disease and the National Scrapie Eradication Program Prepared by the Scrapie Management Team USDA, APHIS, Veterinary Services September, 2012 An Introduction to the National Scrapie Eradication Program This overview provides Federal and State regulatory personnel with a basic introduction to scrapie and the National Scrapie Eradication Program (NSEP). Since the association between BSE in cattle and variant CJD in humans was made in the mid-1990s, a great deal of research on TSE diseases has been done. Recognizing the importance of eliminating TSE diseases from food animal populations, in the past decade many countries around the world – including the United States – have initiated aggressive eradication programs for scrapie. How to Use this Document This introduction to scrapie briefly describes the disease and outlines the major elements of the National Scrapie Eradication Program (NSEP). This document is part of the National Scrapie Reference Library. The Reference Library is a collection of the major documents and templates relevant to the NSEP. Whenever a topic that has been summarized has more extensive information available, the relevant documents in the National Scrapie Reference Library are be referenced so the reader can learn more on the subject. This document has bookmarks for each section and then subsection for easier navigation. If the bookmarks panel is not already activated, click on the bookmarks icon along the upper left-hand side of the screen to open it. The bookmarks icon looks like this: . 1 | Page An Introduction to the National Scrapie Eradication Program Contents Learning Objectives ................................................................................................................................ 4 Section One: History of Scrapie in the United States ............................................................................ -

Course Material and Quiz Answer Form (PDF)
California Association for Medical Laboratory Technology Distance Learning Program PRION DISEASES Course # DL-983 by Rebecca Rosser, MA Education & Development Consultant - Laboratory Kaiser Permanente SCPMG Regional Reference Laboratory North Hollywood, CA Approved for 1.0 CE CAMLT is approved by the California Department of Public Health as a CA CLS Accrediting Agency (#21) Level of Difficulty: Intermediate 39656 Mission Blvd. Phone: 510.792.4441 Fremont, CA 94539 FAX: 510.792.3045 Notification of Distance Learning Deadline DON’T PUT YOUR LICENSE IN JEOPARDY! This is a reminder that all the continuing education units required to renew your license/certificate must be earned no later than the expiration date printed on your license/certificate. If some of your units are made up of Distance Learning courses, please allow yourself enough time to retake the test in the event you do not pass on the first attempt. CAMLT urges you to earn your CE units early! DISTANCE LEARNING ANSWER SHEET Please Circle the one best answer COURSE NAME: PRION DISEASES COURSE # DL-983 NAME ____________________________________ LIC. # ____________________ DATE ____________ SIGNATURE (REQUIRED) ___________________________________________________________________ EMAIL_______________________________________________________________________________________ ADDRESS _____________________________________________________________________________ STREET CITY STATE/ZIP 1.0 CE – FEE: $12.00 (MEMBER) | $22.00 (NON-MEMBER) PAYMENT METHOD: [ ] CHECK OR [ ] CREDIT CARD # _____________________________________ TYPE – VISA OR MC EXP. DATE ________ | SECURITY CODE: ___ - ___ - ___ 1. a b c d 2. a b c d 3. a b c d 4. a b c d 5. a b c d 6. a b c d 7. a b c d 8. a b c d 9. a b c d 10. -
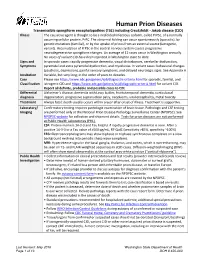
Prion Disease Reporting and Investigation Guideline
Human Prion Diseases Transmissible spongiform encephalopathies (TSE) including Creutzfeldt - Jakob disease (CJD) Illness The causative agent is thought to be a misfolded infectious isoform, called PrPSc, of a normally occurring cellular protein, PrPC. The abnormal folding can occur spontaneously (sporadic), by genetic mutations (familial), or by the uptake of prions from an external source (iatrogenic, variant). Accumulation of PrPSc in the central nervous system causes progressive neurodegenerative spongiform changes. An average of 12 cases occur in Washington annually. No cases of variant CJD have been reported in Washington state to date. Signs and In sporadic cases: rapidly progressive dementia, visual disturbances, cerebellar dysfunction, Symptoms pyramidal and extra pyramidal dysfunction, and myoclonus. In variant cases: behavioral changes (psychosis, depression), painful sensory symptoms, and delayed neurologic signs. See Appendix A Incubation Variable, but very long; in the order of years to decades. Case Please see https://www.cdc.gov/prions/cjd/diagnostic-criteria.html for sporadic, familial, and Classification iatrogenic CJD and https://www.cdc.gov/prions/vcjd/diagnostic-criteria.html for variant CJD. Report all definite, probable and possible cases to CDE Differential Alzheimer’s disease, dementia with Lewy bodies, frontotemporal dementia, corticobasal diagnosis degeneration, progressive supranuclear palsy, neoplasms, viral encephalitis, metal toxicity Treatment Always fatal; death usually occurs within a year after onset of illness. Treatment is supportive. Laboratory/ Confirmatory testing requires pathologic examination of brain tissue. Pathologic and CSF testing Imaging are performed only at the National Prion Disease Pathology Surveillance Center (NPDPSC). See NPDPSC website for collection and shipment details. Tests for prion diseases are not performed at Public Health Laboratories (PHL). -
Cell Science & Stem Cell Research
Yervand E. Karapetyan, J Cell Sci Ther 2012, 3:8 http://dx.doi.org/10.4172/2157-7013.S1.022 2nd World Congress on Cell Science & Stem Cell Research November 12-14, 2012 Hilton San Antonio Airport, USA Long double stranded RNA is present in scrapie infected cells and tissues Yervand E. Karapetyan Oncological Dispensary, Artsakh Republic he nature of the infectious agent causing scrapie and other Transmissible Spongiform Encephalopathies remains an enigma Tdespite decades of research efforts. The protein-only prion hypothesis posits that abnormal conformer of a host protein is the infectious agent. Virus and virino theories include host-independent nucleic acid as the genome of the infectious agent in addition to protein component (in case of virino a host protein and in case of virus a viral protein). Viral or sub-viral nucleic acids have long been sought in scrapie to explain the existence of multiple agent strains. Many different approaches were undertaken to find such nucleic acid. Despite that no scrapie specific nucleic acid sequences have been found in infected cells or tissues. Most viruses induce synthesis of long double stranded RNA (dsRNA) during their replication in cells. Therefore the presence of long dsRNA would be an indication of viral infection in cells. J2 monoclonal antibody against long dsRNA is a great tool for easy screening of cells and tissues for the presence of suspected unknown viral infection. This antibody has not been used for testing of scrapie infected tissues. Evidence is presented here for long dsRNA in scrapie infected cells and tissues. Such dsRNA is also found in scrapie free tissue culture cells.